
Translate this page into:
Primary Hyperoxaluria Type 1: A great masquerader
*Corresponding author: Dr. Chintan G. Shah, Division of Pediatric Nephrology, Department of Pediatrics, Bai Jerbai Wadia Hospital for Children, Mumbai, Maharashtra, India. chintushah41090@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shah CG, Ohri AJ, Udani AH. Primary Hyperoxaluria Type 1: A great masquerade. Wadia J Women Child Health 2022;1(1):13-7.
Abstract
Primary hyperoxaluria (PH) Types I, II, and III is an autosomal recessive inherited disorder of defect in glyoxylate metabolism due to specific hepatic enzyme deficiencies causing renal damage due to deposition of oxalate crystals that induce renal epithelial cell injury, and inflammation resulting in reduced renal oxalate elimination leading to extra renal deposition of calcium oxalate crystals. PH is under diagnosed because of phenotypic heterogeneity masquerading as infantile nephrocalcinosis (NC) with or without renal failure or renal calculus disease in adults. We present three children with genetically proven PH1 seen over last 2 years along with a brief review of the literature. In this series all cases were female. Two girls had infantile onset of symptoms and one presented in childhood. Renal failure in all with varying sonography features including small size kidneys, multiple renal calculi, bulky kidneys with loss of corticomedullary differentiation were seen. Extrarenal affection was seen in one child. Renal replacement therapy was provided in all. Awareness of PH and early diagnosis by measurement of plasma and urinary oxalate and molecular characterization helps in prompt aggressive therapy, preventing extrarenal manifestations and plan long term management.
Keywords
Primary hyperoxaluria
Nephrocalcinosis
Renal replacement therapy
Molecular characterization
Molecular subtyping

INTRODUCTION
Primary hyperoxalurias (PH) are a rare, autosomal recessive inherited, group of inborn errors of metabolism characterized by excessive hepatic production of oxalate. There are three main types that PH1 caused by a deficiency of the liver specific peroxisomal enzyme alanine/glyoxylate aminotransferase (AGXT), PH2 caused by a deficiency of cytosolic enzyme glyoxylate reductase/ hydroxy pyruvate reductase and PH3 is linked to the gene HOGA1 encoding the mitochondrial enzyme 4-hydroxy-2-oxoglutarate aldolase. PH1 is the most severe form of PH accounting for about 80% of genetically characterized cases. In the current study, we aim to highlight the spectrum of clinical presentation of PH1 using high index of suspicion and utilizing the available diagnostic tools for proper clinical phenotyping. Suspected cases on clinical grounds were further confirmed by genetic mutational analysis.
MARTIAL AND METHODS
Retrospective observational study was carried out at Division of Pediatric Nephrology, Bai Jerbai Wadia Hospital for children, Mumbai. Records of children diagnosed as PH1 over 2 years period were studied for demographic details, clinical features and laboratory profile at last presentation.
Laboratory work up done as per unit protocol, included complete blood count, renal function test, serum values for calcium, phosphorous, uric acid, and urine analysis. Plain x ray abdomen was done in suspected renal calculus disease. Ultrasound KUB done to identify renal morphology and calcifications was recorded. 24 h urine metabolic profile including oxalate was done. If 24 h urinary oxalate excretion exceeded the upper-limit (<45 mg/1.73 m2). PH was the most likely diagnosis. Renal biopsy done as indicated was studied for features of PH that showed birefringence on polarized microscopy suggesting oxalate deposits characteristic of PH. All the patients with clinico-laboratory parameters suggestive of PH were counselled to undergo genetic analysis for diagnostic confirmation.
Therapy included peritoneal/hemodialysis, supportive therapy for chronic kidney disease (CKD), oral pyridoxine, capsule oxalobacter formigenes as indicated.
Case 1
Six months old male born of 2° consanguinity, delivered normally at 38 week Gestational Age of an uneventful pregnancy, was admitted with an afebrile episode of profuse watery loose stools, vomiting and decreased urine output. Laboratory investigations revealed raised serum creatinine (5.8 mg/dl), severe metabolic acidosis, hyperkalemia and anemia [Table 1]. Renal sonography showed bilateral bulky kidneys with increased echogenicity with loss of corticomedullary differentiation (CMD). Peritoneal dialysis was performed and supportive management as appropriate was given. In view of unexplained renal failure, renal biopsy was performed which showed 15 glomeruli. On histopathology most of the proximal tubules showed intraepithelial and intraluminal oxalate crystals with denudation of tubular epithelial cells. Polarized microscopy showed brightly birefringent crystals [Figure 1]. Diagnosis of PH (Type 1) was made and molecular characterization showed a homozygous missense variation in exon 6 of the AGXT gene (chr2:g.240873996C>T; Depth: 159x) that results in the amino acid substitution of Leucine for Serine at codon 205 (p.Ser205Leu; ENST00000307503.4) [Table 2]. The child now 16 months age, is on continuous ambulatory peritoneal dialysis.
| Laboratory investigations | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Hemoglobin (g/dl) | 7.9 | 8.2 | 7.6 |
| BUN (mg/dl) | 98 | 102 | 131 |
| Serum creatinine (mg/dl) | 5.1 | 5.2 | 7.8 |
| Serum sodium (mEq/L) | 125 | 131 | 127 |
| Serum potassium (mEq/L) | 6.0 | 5.9 | 6.2 |
| pH | 7.2 | 7.1 | 7.1 |
| Serum bicarbonate | 8.1 | 8.2 | 6.9 |
| Case No. | Age of presentation | Gene | Location | Variant | Zygosity | Inheritance | Classification |
|---|---|---|---|---|---|---|---|
| 1. | 6 months | AGXT (+) (ENST00000307503.4) | Exon 6 | c. 614C>T (p.Ser205Leu) | Homozygous | Autosomal recessive | Likely Pathogenic |
| 2. | 6 months | AGXT (+)(ENST00000307503.3) | Exon 4 | c. 447_454del (p.Leu151AsnfsTer14) | Homozygous | Autosomal recessive | Pathogenic |
| 3. | 7 years | AGXT (+) (ENST00000307503.3) | Exon 2 | c. 346G>A (p.Gly116Arg) | Homozygous | Autosomal recessive | Likely Pathogenic |
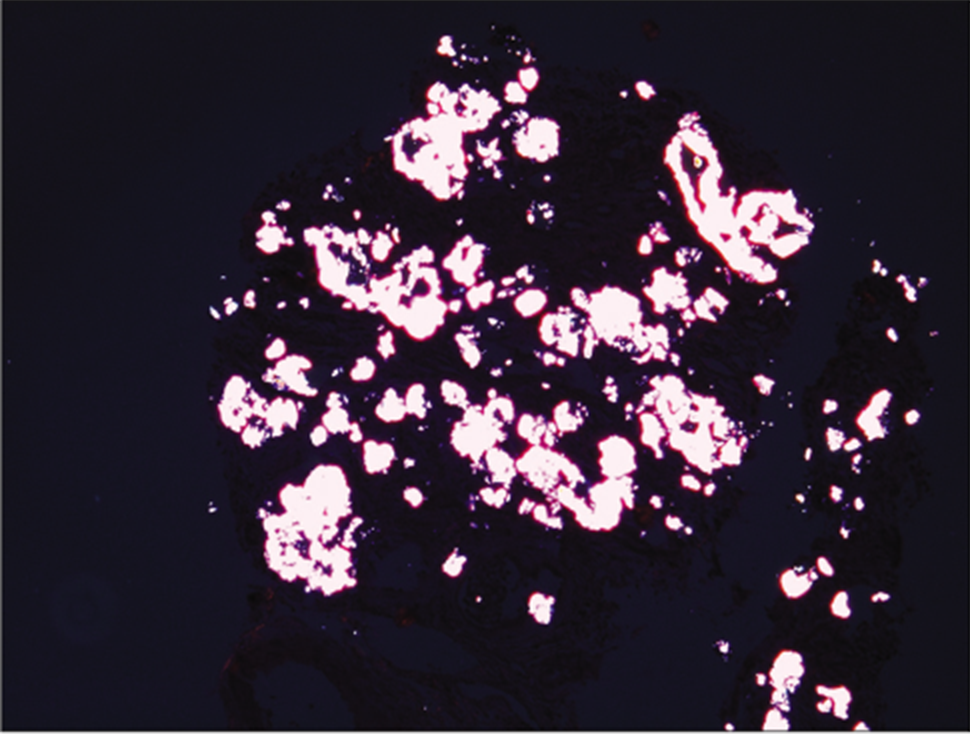
- Polarized microscopy showing birefringence appearance.
Case 2
Three years old female, born of non-consanguineous union presented with growth failure noticed from 6 months of age and a suspicion of auditory inattention. At presentation her weight was 8.2 kg (< 3rd centile) and height of 74 cm (<3rd centile). She was pale with normal blood pressure. Laboratory investigations revealed severe metabolic acidosis, raised serum creatinine (5.1 mg/dl), hyperkalemia and anemia [Table 1]. Renal sonography showed bilateral small size kidneys with increased echogenicity and loss of CMD. Bilateral profound sensorineural hearing loss was detected on audiometry. Ophthalmologic evaluation was suggestive of multiple hypopigmented deposits in macula with mild arteriolar attenuation with no evidence of lenticonus. As kidney sizes were small, kidney biopsy was not done. Molecular analysis revealed homozygous 8 base pair deletion in exon 4 of the AGXT gene (chr2:g.241810789_241810796del; Depth: 228x) that results in a frameshift and premature truncation of the protein 14 amino acids downstream to codon 151 (p.Leu151AsnfsTer14; ENST00000 30750 3.3) causing PH1 [Table 2] She was put on conservative management of CKD. Presently at the age of 5 years, her weight is 14.2 kg (10th centile) and height 97.4 cm (3rd centile). She is in CKD Stage V on conservative management and pre-emptive combined liver kidney transplant is being planned.
Case 3
Seven year old female admitted with complaints of severe pain in abdomen, vomiting and anuria. In view of anuria with highly raised serum creatinine (7.8 mg/dl) and uremic symptoms, prompt haemodialysis was initiated. Plain X-ray abdomen [Figure 2] and renal sonography revealed bilateral gross hydronephrosis with multiple mobile calculi within both renal pelvis, bilatreral echogenic kidneys with loss of CMD, right ureteric calculi 1 cm, left upper and mid ureter multiple calculi. There was a strong family history of kidney stones in first degree relatives. Twenty four hour urinary calcium was 2.4 mg/kg/day (normal - <4 mg/kg/day), 24 h urinary oxalate was raised at 75mg/1.73m2/day (normal- <45 mg/1.73 m2/day). Child underwent left pyelolithotomy with urterolithotomy and 18 calculi were removed. Stone analysis was suggestive of calcium ammonium oxalate stones. Urine output got established after procedure and creatinine started decreasing. Child did not require further haemodialysis and was discharged with serum creatinine of 1.1 mg/dl on tablet vitamin B6, capsule oxalobacter formigenes. Child was non compliant with medications and was readmitted 6 months later with similar complaints of pain abdomen and a serum creatine level of 12mg/dl. This time she required maintenance haemodialysis for long and gradually shifted to haemodialysis 3 times per week through a permanent catheter and transferred back to her district hometown. She is currently on regular haemodialysis. Molecular studies revealed a homozygous missense variation in exon 2 of the AGXT gene (chr2:g.241808767G>A; Depth: 259x) that results in the amino acid substitution of Arginine for Glycine at codon 116 (p.Gly116Arg; ENST00000307503.3) was detected suggestive eof PH1.
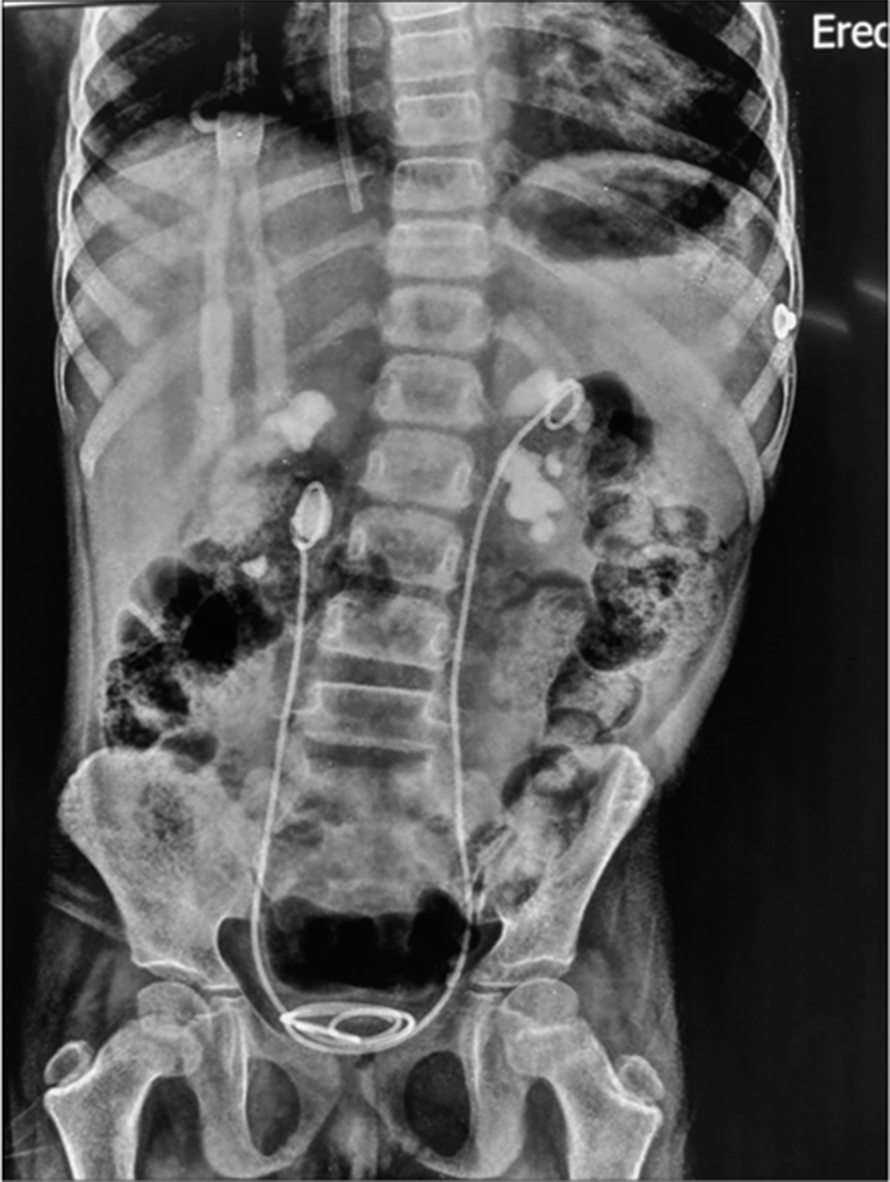
- Multiple stones in the urinary tract with bilateral DJ stent in situ.
DISCUSSION
PH1 is an autosomal recessive disorder caused by a deficiency of the liver-specific peroxisomal enzyme alanine-glyoxylateaminotransferase (AGXT). AGXT is a pyridoxal-phosphate (PLP) - dependent liver-specific peroxisomal enzyme which catalyzes the transamination of the intermediary metabolite glyoxylate to glycine. In PH1, AGXT deficiency allows the glyoxylate to diffuse through the peroxisomal membrane into the cytosol where it is oxidized to oxalate. With advancing renal failure plasma oxalate rises. Oxalosis is a state when above a plasma oxalate concentration of 50 mmol/l, the solubility of calcium oxalate in plasma is exceeded,[1] and extrarenal deposition of oxalate in tissues like bone, retina, myocardium, and medium size and major arteries begins.
PH1 is estimated to account for 1–2% of pediatric end stage renal disease (ESRD) population in registries from USA, UK and Japan.[2] Ten percent of Kuwaiti children and 13% of Tunisian children with ESRD are reported to have PH1.[3] PH1 shows considerable phenotypic, enzymatic and genotypic heterogeneity. In our study uremia was the commonest renal manifestation seen in all cases as compared to Von Woerden et al. who reported only in 40% of their cases.[4] In a descriptive cohort study by Soliman et al., which included patients with presumable (why are you calling it presumable) PH1, seventeen cases (65.4%) had reached end-stage renal disease (ESRD): 6/17 (35.3%) during infancy, 4/17 (23.5%) in early childhood and 7/17 (41.29%) in late childhood. Two cases demonstrated extrarenal manifestations of oxalosis.
A female with infantile onset PH1, had visual impairment associated with nystagmus and subnormal retinal function. She also had myocardial deposits diagnosed by echocardiography and confirmed by cardiac MRI. Another case had severe bone disease and bone marrow failure with oxalate deposits diagnosed in bone marrow aspirate.[5] In our series of 3 cases, we had infantile and childhood onset of symptoms. One child had extrarenal manifestations in the form of retinal deposits and profound sensorineural hearing loss. These extrarenal manifestations are largely preventable by early diagnosis and aggressive renal replacement therapy when indicated. In a study by Kapadia et al., 13 of 15 patients were identified to have genetically confirmed PH. Three (37.5%) out of eight PH1 patients had more than one extra renal involvement in the form of cardiovascular dysfunction (2), skeletal osteopathy (1), retinal involvement (1) and recurrent cholangitis-pancreatitis (1).[6]
Many point mutations in the AGXT gene encoding the enzyme have been identified in PH1. There is poor correlation between the genotype, degree of enzyme deficiency and clinical severity of PH1.[7] In a study by Kathrina et al., PH1 cohort displayed many characteristics reported in smaller cohorts; two AGXT variants,mutant alleles (p.G170R [32.3%] and c.33dupC [15.5%]), accounted for 47.8% of PH1, and most mutations were missense, even after exclusion of common alleles. In our series, missense mutation in 2 cases and truncation in 1 was seen. In a study by Williams et al. on selected exon sequencing of the AGXT gene, of all the novel mutations identified, c.447_454delGCTGCTGT was the most common and was detected exclusively in association with the major allele in 6 unrelated patients, all of whom were homozygous and of Asian origin.[8] In Indian literature, Chanchalani et al. reported 3 patients of PH1 and sequencing of AGXT gene revealed a missense homozygous mutation in exon two (c.302T > C), which is expected to form proline instead of leucine (Leu101Pro) on translation.[9] AGXT gene mutation were found in 8 (61.54%) out of 13 patients by Kapadia et al. 75% patients had homozygous and 25% had compound heterozygous mutations. As per ACMG criteria 5 (62.5%) mutations were classified as pathogenic, 3 (27.5 %) as likely pathogenic.[6]
When assessing the relevance of various types of mutations as possible therapeutic targets, we should consider allele frequency in various populations and what type of mutation is correctable. Insertion-deletion (except for cases of short in-frame mutations) and splice site aberrations are not correctable, since they do not result in a full-length protein. Patients with null mutations, such as c.447_454delGCTGCTGT, which produce no AGT protein, may be unresponsive to pyridoxine therapy and hence be more likely to require liver transplant for effective treatment.[10] However, these types of mutations, with the exception of c. 33dupC, are infrequent among PH1 patients.
Nonsense mutations may be subjects for read-through therapeutic approaches. The most abundant AGXT nonsense mutation is R122X. Yet, missense mutations are most prevalent among PH1 patients and a few of them together are responsible for about 80% of all PH1 cases.
Fargue et al. have shown intracellular PLP appears to work as a chemical chaperone (increasing net expression and peroxisomal import) of the most common mistargeted mutant form of AGXT (i.e. Gly170Arg on the background of the polymorphic minor allele). In the first prospective study published by Hoyer-Kuhn et al., 6 out of 12 patients showed over 30% reduction in urinary oxalate levels but none achieved complete biochemical remission.[10]
Oxalate degradation by Oxalobacter formigenes involves three unique proteins, an oxalate: formate membrane transporter, oxalylCoA decarboxylase, and formyl-CoA transferase.[11] In a study by Hoppe et al. Oxalobacter was orally administered for 4 weeks as frozen paste (IxOC-2) or as enteric-coated capsules (IxOC-3). Urinary oxalate or plasma oxalate in renal failure was determined at baseline, weekly during treatment and for a 2-week follow-up. The patients who showed ≥20% reduction in both urinary and plasma oxalate level at the end of weeks 3 and 4 were considered as responders.[12]
The modality of choice is intensified daily haemodialysis as oxalate clearance in peritoneal dialysis (PD) is inefficient.[13] A combination of daily nocturnal peritoneal dialysis with haemodialysis has also been suggested. Unfortunately, there is no dialysis strategy that can offset oxalate generation by the liver.[1] Combined liver and kidney transplantation prevents disease recurrence in the kidney allograft, as liver transplantation with removal of the native liver corrects the metabolic defect.[14] The 5-year survival of children with PH undergoing isolated kidney or combined liver kidney transplantation is 14% and 76%, respectively. In order to prevent oxalate-induced damage to the kidney allograft, KDIGO guidelines recommend urinary dilution with aggressive hydration and frequent dialysis for patients with delayed graft function, in the immediate post-transplantation period.[14]
Therapeutic approaches that are currently under development are substrate reduction therapy (SRT), Replacement therapy (gene therapy, cell therapy, and enzyme replacement therapy), chaperone-proteostasis regulator therapy and anti-inflammatory therapy. PH1 individuals with preserved kidney function or moderate CKD will be benefited from RNAi-based SRTs that are currently undergoing advanced phases of clinical trials and will hopefully soon be approved by the regulatory authorities.[14]
CONCLUSION
PH1 is one of the most challenging diseases being highly heterogeneous with variable age of first presentation and vague initial symptoms that can easily result in diagnostic delay. Moreover, lack of awareness and the low index of clinical suspicion play a major role in PH1 under-diagnosis. Family history of stone disease/ESRD, radio-opaque nephrolithiasis and/or NC, cortical and medullary NC mostly misinterpreted as simply “hyperechogenic” kidneys in infants and toddlers are helpful clues to diagnosis that need to be enforced by the availability of 24 h urine oxalate, renal biopsy and further confirmed by molecular genetic analysis. The application of Next Generation Sequencing (NGS) will hopefully shortly allow the detection of additional genes that, in their mutated form, lead to increased oxalate production and excretion.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Serum calcium oxalate saturation in patients on maintenance haemodialysis for primary hyperoxaluria or oxalosis-unrelated renal diseases. Clin Sci (Lond). 1991;81:483-90.
- [CrossRef] [PubMed] [Google Scholar]
- The 1998 report of the Japanese National Registry data on pediatric end-stage renal disease patients. Pediatr Nephrol. 2002;17:456-61.
- [CrossRef] [PubMed] [Google Scholar]
- Primary hyperoxaluria the German experience. Am J Nephrol. 2005;25:276-81.
- [CrossRef] [PubMed] [Google Scholar]
- Primary hyperoxaluria Type 1 in Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003;18:273-9.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical spectrum of primary hyperoxaluria Type 1: Experience of a tertiary center. Nephrol Ther. 2017;13:176-82.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and genetic profile of patients with primary hyperoxaluria: Observation from a single centre from West India. Int J Contemp Pediatr. 2021;8:451-9.
- [CrossRef] [Google Scholar]
- Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26:2559-70.
- [CrossRef] [PubMed] [Google Scholar]
- Selected exonic sequencing of the AGXT gene provides a genetic diagnosis in 50% of patients with primary hyperoxaluria Type 1. Clin Chem. 2007;53:1216-21.
- [CrossRef] [PubMed] [Google Scholar]
- Common mutation underlying primary hyperoxaluria Type 1 in three Indian children. Indian J Nephrol. 2012;22:459-61.
- [CrossRef] [PubMed] [Google Scholar]
- Vitamin B6 in primary hyperoxaluria I: First prospective trial after 40 years of practice. Clin J Am Soc Nephrol. 2014;9:468-77.
- [CrossRef] [PubMed] [Google Scholar]
- Cloning, sequencing, and expression in Escherichia coli of OxlT, the oxalate: Formate exchange protein of Oxalobacter formigenes. J Biol Chem. 1996;271:6789-93.
- [CrossRef] [PubMed] [Google Scholar]
- Oxalobacter formigenes: A potential tool for the treatment of primary hyperoxaluria Type 1. Kidney Int. 2006;70:1305-11.
- [CrossRef] [PubMed] [Google Scholar]
- European PHI transplantation study group 2005 a 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): The European PH1 transplant registry experience 1984-2004. Am J Nephrol. ;39:282-9.
- [CrossRef] [PubMed] [Google Scholar]
- KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(Suppl 3):S1-155.
- [CrossRef] [Google Scholar]




