
Translate this page into:
A rare case of polysplenia syndrome associated with atrioventricular canal defect and type four ileal atresia in a preterm neonate: A case report and literature review
*Corresponding author: R. R. Prashanth, Department of Neonatology, Seth Gordhandas Sunderdas Medical College (GSMC) and KEM Hospital, Mumbai, Maharashtra, India. prash2635@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Prashanth RR, Jyothi S, Haribalakrishna A. A rare case of polysplenia syndrome associated with atrioventricular canal defect and type four ileal atresia in a preterm neonate: A case report and literature review. Wadia J Women Child Health. 2024;3(1):35-9. doi: 10.25259/WJWCH_3_2024
Abstract
Antenatal diagnosis of heterotaxy syndrome is only the tip of the iceberg. These neonates can have a myriad of multi-system anomalies, each of which requires anticipation and an individualized management plan. The spectrum of heterotaxy syndrome ranges from incidentally detected innocuous anomalies to major life-threatening congenital malformations such as atrioventricular canal defects and bowel atresia. Neonatal sepsis tends to be more fulminant and catastrophic in these neonates with complex anomalies and poor outcomes that necessitates better-informed antenatal counseling regarding the termination of such pregnancies.
Keywords
Polysplenia
Heterotaxy
Prematurity
Neonate
Ileal atresia
Atrioventricular canal defect

INTRODUCTION
Establishment of the normal axis of the human body results in an orderly arrangement of all organs. This phenomenon constitutes the formation of the human body and has always fascinated researchers all over the world. A lot of these fine mechanisms remain yet to be revealed. Most of the molecular genetic process influencing axes formation takes place before and during gastrulation phase.[1] Defective development of this can result in a wide spectrum of anomalies with an array of clinical presentations ranging from incidentally detected asymptomatic anomalies to life threatening lethal congenital malformations. Such defective development results in heterotaxy syndromes which are characterized by abnormal arrangement and morphology of abdominal and thoracic organs.[2] In this case report, we describe a rare case of polysplenia syndrome with complete atrioventricular (AV) canal defect, type 4 ileal atresia and situs ambiguous with liver in midline, stomach on the right side along with two spleens.
CASE REPORT
A 21-year-old primigravida mother at 35 weeks of gestation was referred to our hospital in spontaneous preterm labor. Her second and third trimester scans revealed grossly abnormal structural malformations. Second trimester scan suggested the possibility of truncus arteriosus while the third trimester scan showed abnormal position of liver on the left side, raising the suspicion of a heterotaxy syndrome. No additional antenatal counseling or tests were done. There was no significant family history of similar illness.
A male neonate weighing 1968 gram, was born by normal vaginal delivery within few hours of onset of labour. Baby had APGAR scores of 9 at both 1 and 5 min, and no resuscitation was required at birth. Examination at birth showed a head circumference of 30.5 cm, total length of 42 cm, heart rate - 128/min, respiratory rate - 58/min, SpO2 (oxygen saturation) 92%. Capillary refill time (CRT) <3 seconds, and Silverman Anderson Score (SAS) - 0/10. Femoral pulses were well felt, the liver was palpable 2 cm below the left costal margin, and the spleen was palpable 1 cm below the right costal margin. Neurologically, the cry, tone, and reflexes were normal. He was hemodynamically stable and was shifted to neonatal intensive care unit (NICU) for further management and was started on complete milk feeds with a cup and spoon.
Following the first feed, within 3 hours of NICU admission, the neonate developed large volume bilious aspirates and abdominal distension with multiple episodes of apnea. Sepsis screen was negative, there was no evidence of shock clinically and on echocardiogram. He was started on non-invasive ventilation with minimal settings and kept nil per oral. An urgent X-ray chest and abdomen was done which showed cardiomegaly, a right-sided stomach with dilated bowel loops and absence of rectal gas shadow but no evidence of pneumonia. The absence of rectal gas shadow persisted in subsequent X-rays [Figure 1]. While the neonate was being planned for surgical exploration in view of suspected surgical pathology, further investigations were done to rule in other major congenital anomalies.
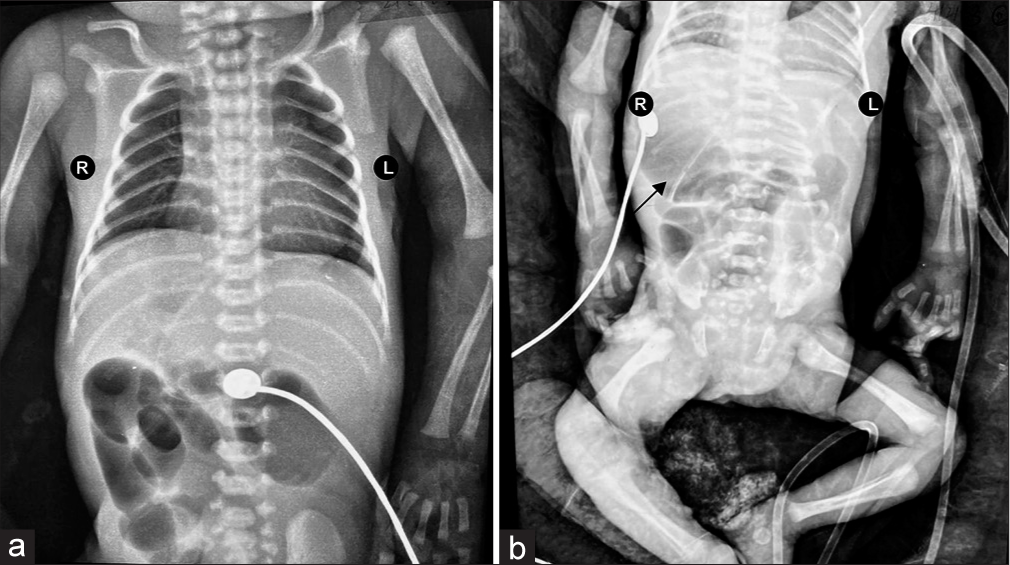
- (a) X-ray chest and abdomen AP view showing cardiomegaly with a cardiothoracic ratio of 0.6. (b) Supine X-ray abdomen showing right-sided stomach (black arrow) and absent rectal gas shadow. AP: Antero-posterior.
Bed side ultrasonography (USG) suggested a midline liver and spleen was not visualized. 2D echo was suggestive of levocardia with complete AV canal defect with common atrium and common AV valve with mild regurgitation. There was a large inlet ventricular septal defect (VSD) and a moderate sized patent ductus arteriosus with left to right shunt. The great arteries were normally related and biventricular function was normal.
Exploratory laparotomy done on the 5th day of life revealed a type 4 Ileal atresia (string of sausage appearance), approximately 40 cm from the duodenum- jejunal junction [Figure 2]. Contrary to the USG abdomen which could not locate the spleen, laparotomy revealed the presence of 2 small sized spleens on the right side [Figure 3]. Liver was predominantly in the midline extending to the left. The large intestine was not fixed to the abdominal wall and the caecum was found on the left side. Divided ileostomy was done and the baby was shifted back to the NICU in an intubated state.
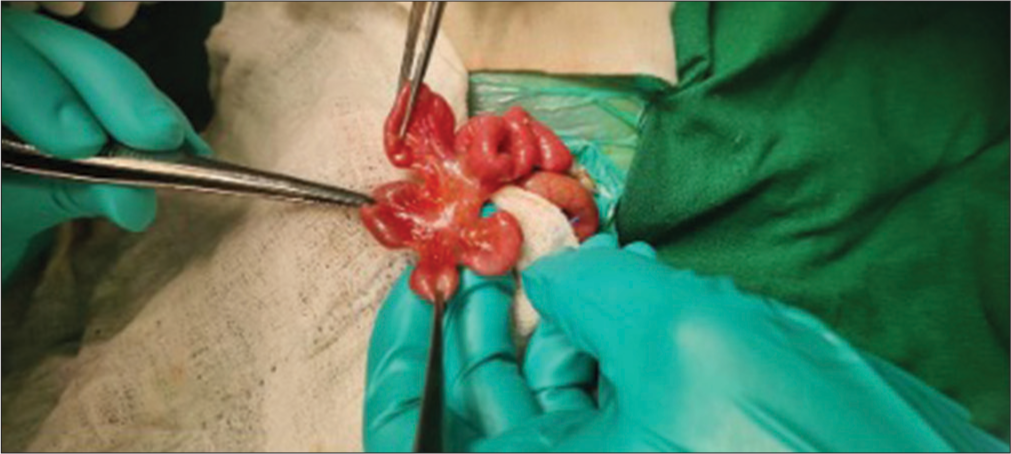
- Type 4 ileal atresia (string of sausage appearance).
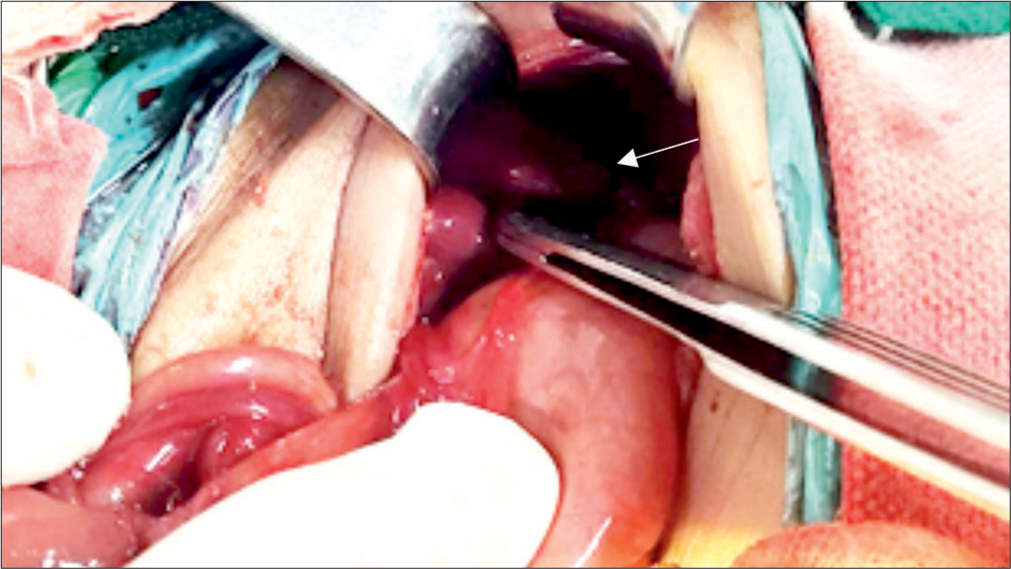
- Polysplenia found during laparotomy- 2 small-sized spleen on the right side (white arrow).
In the post operative period, the neonate developed ileus which persisted for 6 days during which he was on total parentral nutrition. Enteral nutrition was introduced on 7th post-operative day (POD). We succeeded in extubating the neonate on POD 8 and feeds were gradually increased and were well tolerated. On POD 20, the baby developed culture negative late onset neonatal sepsis and consumptive coagulopathy followed by a rapid deterioration in the clinical status requiring mechanical ventilation and broad-spectrum antibiotics. Repeat echocardiography showed no evidence of worsening underlying cardiac pathology. The neonate later developed refractory septic shock and succumbed as a result of pulmonary hemorrhage on POD 25. Karyotype was normal. An autopsy consent was sought to find out the extent of congenital anomalies and the exact cause of death, but the parents did not consent.
DISCUSSION
Situs solitus represents the normal arrangement of abdominal and thoracic organs with levocardia, liver, trilobed lung on the right and stomach, and bilobed lung on the left. Situs inversus is the mirror image of the above mentioned normal arrangement with dextrocardia, liver on the left and stomach on the right side. Situs ambiguous is a disordered arrangement of organs that is neither a solitus nor an inversus pattern. The terms heterotaxy syndromes, cardiosplenic syndromes and right or left isomerism have been used interchangeably for these disorders.
Isomerism or the abnormal symmetry of normally asymmetric viscera is a salient feature of heterotaxy syndromes. Conventionally, left isomerism is characterized by the presence of paired left viscera with well documented congenital cardiac anomalies. Hence, a left isomerism (polysplenia syndrome) indicates the presence of bilateral bilobed lungs, bilateral left atrial appendages, multiple splenules, an abnormal position of liver and associated characteristic congenital malformations such as interrupted inferior vena cava (IVC) with azygos continuation. A right isomerism (asplenia syndrome and Ivemark syndrome) is associated with bilateral right atrial appendages, bilateral trilobed lung, absent spleen, and a malpositioned IVC anterior or juxtaposed to aorta.
Complete atrioventricular septal defect (AVSD) though documented in both right and left isomerism, is reported to be more commonly associated with right isomerism.[3] The common cardiac findings in the left isomerism are bilateral superior vena cava, ventricular septal defect (VSD), partial anomalous pulmonary venous connection, truncus arteriosus and congenital heart block. Besides AVSD, in right isomerism total anomalous pulmonary venous connections, transposition of great arteries, and double outlet right ventricle are seen. The diagnostic criteria in infants for heterotaxy syndromes mandates the presence of complex congenital heart disease in addition to any 2 of the following anomalies i.e IVC and abdominal aorta on the same side, interrupted IVC, isomerism of atrial appendages, isomerism of lung lobes, polysplenia or asplenia, inverted or symmetrical liver and stomach.[3] The most common constellation of findings described in polysplenia syndrome are AVSD, IVC defects, bilateral bilobed lungs, stomach on right side, liver in the midline, and various degrees of intestinal malrotation.[1] Polysplenia refers to the presence of two or more spleens. Polysplenia has been found to be associated with many viscero-cardiac anomalies associated with heterotaxy, collectively known as polysplenia syndromes.
The incidence of the heterotaxy syndromes range from 1 to 1.5/10,000 live births and is associated with a very high mortality rate.[4] Long-term outcomes of these syndromes are usually determined by the severity of the cardiac anomalies.[5] The syndrome is also associated with many congenital malformations of gastro intestinal tract, such as biliary atresia, intestinal atresia, volvulus, and varying degrees of bowel malrotation which also influence the long-term morbidity, mortality, and survival of these babies.[3]
The heart initially develops as a straight tube, which undergoes folding in an orderly fashion to eventually develop into a four chambered organ, which contracts and relaxes in a synchronized manner. Development of endocardial cushions, septation of conotruncus and alignment of inflow and outflow tracts is established between 28 and 35 days of gestation, which coincides with the period of development of spleen from left mesogastrium.[6-8] An insult to the embryo at this crucial juncture has been postulated as the mechanism behind laterality defects. Molecular genetics has also explained the mechanisms behind the defective embryogenesis in heterotaxy syndromes. The break in symmetry during embryogenesis occurs as a result of the characteristic pattern of rotation of nodal cilia. This causes a left-ward nodal flow resulting in generation of signals facilitating asymmetry which are transmitted to left lateral plate mesoderm. These activate the left side specific growth and transcription factors (Nodal, Lefty2, and Pitx2c) that trigger the development of left sided structures while the right sided structures develop from right lateral plate by default. Hence, any dysfunction of these transcription factors could result in defective organogenesis, heterotaxy, and associated congenital cardiac malformations. Some of the genes which are associated with heterotaxy syndromes are ZIC3, NODAL, CFC1, LEFTY2 and GDF1.[4] In addition to these genetic factors, many studies have observed a role for many environmental disruptors during early embryogenesis in the etiology of these defects. Important factors postulated to have a causative role are maternal diabetes, family history of malformations and cocaine abuse by the mother during the first trimester.[9]
During our literature review, we came across a few well documented cases of polysplenia syndromes which had diverse findings in thoraco-abdominal organs, similar to ours. In the case reported by Mohor et al.,[1] polysplenia was found on autopsy associated with a host of other rare findings namely complete atrioventricular (AV) canal defect, bilobed lungs, situs ambiguous with right sided stomach, midline liver, gall bladder agenesis, and complete inversion of liver and pancreas. The patient had developed fulminant Escherichia coli sepsis, clotting disorder and acute renal failure. Huseynova et al [2] reported a case where asplenia was suspected as spleens were not detected in USG and laparotomy and on computed tomography showed 5 splenules in the right upper quadrant of abdomen were seen. This child had associated hypoplastic left heart syndrome, complete gastric volvulus, malrotation of duodenum, multiple jejunal atresia (sausage bowel), centrally located liver and right-sided stomach. Although this child survived post laparotomy, and was weaned off respiratory support, the final outcome remained poor as cardiac surgery was not performed in anticipation of the overall poor prognosis and the patient was placed in palliative care.
Case reported by Rasool and Mirza,[10] mentions polysplenia (7 spleens) diagnosed by laparotomy associated with situs inversus and type 1 jejunal atresia. This case was peculiar with the absence of complex congenital heart disease, and the patient is well at subsequent follow-up. Garcia-Rodriguez et al.[11] described a case of polysplenia syndrome with 13q deletion and associated hypoplastic left heart syndrome, right pulmonary sequestration, diaphragmatic hernia, and pancreatic agenesis. Escobar-Diaz et al.[12] concluded that the 5-year survival rate for polysplenia syndrome was higher at 86% compared to asplenia syndrome which is 53%. The predictors of poor outcome in polysplenia syndrome according to the authors was the presence of univentricular circulation and the left ventricular circulation. Berg et al [3] in their study of the prenatal diagnosis of cardio-splenic syndromes documented no significant differences in severe cardiovascular malformations between the right or left isomerism.
Invariably, the most important step in the management of such cases is accurate prenatal diagnosis. In our case, the mother had a history of erratic antenatal follow-up. Early antenatal scan and timely detection of the anomaly could have facilitated an informed decision making by the parents regarding continuation of pregnancy. As very limited number of cases have been identified and documented so far, there is a possibility of this prenatal diagnosis to be under-prioritized. However, the single case we have managed at our center has taught us otherwise.
CONCLUSION
Although heterotaxy syndromes are a spectrum, the cases that are associated with multiple congenital anomalies, as in ours, are invariably associated with very poor prognosis. We hope that our case report may guide and encourage doctors treating such neonates to anticipate issues, make wiser decisions, and offer better treatment to these unfortunate patients.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- A rare case of polysplenia syndrome associated with severe cardiac malformations and congenital alveolar dysplasia in a one-month-old infant: A complete macroscopic and histopathologic study. J Cardiovasc Dev Dis. 2022;9:135.
- [CrossRef] [PubMed] [Google Scholar]
- Polysplenia syndrome with complex heart disease and jejunal atresia with malrotation in neonate: A case report. Clin Case Rep. 2020;8:848-51.
- [CrossRef] [PubMed] [Google Scholar]
- Prenatal diagnosis of cardiosplenic syndromes: A 10-year experience. Ultrasound Obstet Gynecol. 2003;22:451-9.
- [CrossRef] [PubMed] [Google Scholar]
- The heterotaxy syndrome: Associated congenital heart defects and management. Indian J Thorac Cardiovasc Surg. 2021;37(Suppl 1):67-81.
- [CrossRef] [PubMed] [Google Scholar]
- Intrauterine diagnosis of heterotaxy syndrome. Am Heart J. 2002;143:1002-8.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics. 1988;82:698-706.
- [CrossRef] [PubMed] [Google Scholar]
- Familial clustering of situs inversus totalis, and asplenia and polysplenia syndromes. Am J Med Genet. 1983;16:43-7.
- [CrossRef] [PubMed] [Google Scholar]
- Risk factors for heart disease associated with abnormal sidedness. Teratology. 2002;66:242-8.
- [CrossRef] [PubMed] [Google Scholar]
- Polysplenia syndrome associated with situs inversus abdominus and type I jejunal atresia. APSP J Case Rep. 2011;2:18.
- [Google Scholar]
- A new observation of 13q deletion syndrome: Severe undescribed features. Genet Couns. 2015;26:213-7.
- [Google Scholar]
- Perinatal and infant outcomes of prenatal diagnosis of heterotaxy syndrome (asplenia and polysplenia) Am J Cardiol. 2014;114:612-7.
- [CrossRef] [PubMed] [Google Scholar]




