
Translate this page into:
Spinal neurofibromas presenting as quadriparesis in a case of neurofibromatosis Type 2
-
Received: ,
Accepted: ,
How to cite this article: Kulkarni S, Joshi M. Spinal neurofibromas presenting as quadriparesis in a case of neurofibromatosis Type 2. Wadia J Women Child Health 2022;1(1):24-6.
Abstract
Neurofibromatosis Type II (NF2) is a neurocutaneous syndrome with autosomal dominant inheritance caused by mutation in NF2 gene that codes for protein merlin. It is associated with various tumours such as vestibular schwannoma, meningiomas, ependymomas, etc. in central and peripheral nervous system. The Manchester criteria is used for its diagnosis. We report a case of a 9 year old girl with café au lait spots and plexiform neurofibromas who presented with gradual onset UMN type of progressive quadriparesis over 20 days. Magnetic resonance imaging brain and spine revealed bilateral vestibular schwannomas and multiple spinal neurofibromas which were confirmed on histopathologic examination, thereby confirming diagnosis of NF Type 2.
Keywords
Neurofibromatosis Type 2
Neurofibroma
Meningioma
Vestibular schwannoma

INTRODUCTION
The neurofibromatosis are a group of familial tumour predisposition syndromes characterized by the development of various neoplastic and dysplastic lesions predominantly affecting the central and peripheral nervous systems.[1] Neurofibromatosis Type II (NF2) results from loss-of-function mutations in the NF2 gene on chromosome 22 which codes for a protein product named merlin. NF2 is inherited in autosomal dominant and is most commonly associated with the development of bilateral vestibular schwannomas which are considered pathognomonic. However, patients also have a predisposition to develop other tumors including meningiomas and ependymomas presenting as neurological deficits, hearing loss or peripheral neuropathies.[2] The Manchester Criteria is used for its diagnosis. Management of NF2-associated lesions primarily consists of surgical resection and treatment of symptoms.
CASE REPORT
A 9-year-old girl presented with complaints of bilateral lower limb weakness for 20 days, bilateral upper limb weakness since 10–12 days and neck pain for 7 days. Due to lower limb weakness, she had difficulty in walking and was not able to get up from sitting position. The weakness was equal in both the legs and was progressively increasing since the last 20 days. She was not able to lift her hands above her head and complained of a weak grip. The weakness was more in the left hand and had progressively worsened over 10–12 days. The child also complained of a dull aching pain at the back of the neck of mild to moderate intensity for the last 7 days which was associated with reduced range of motion. There was no history of seizures, headache, vomiting, bowel and bladder involvement and visual and speech disturbances. The cranial nerves and sensory system appeared to be uninvolved. There was no family history of similar complaints. On examination, the child had 5 café au lait spots on the trunk and limbs all measuring more than 15mm and multiple plexiform neurofibromas [Figures 1 and 2]. The tone was normal in all limbs without any visible wasting of muscles. The muscle power was 4/5 across all muscle groups except both shoulders where it was 3/5. Weakness of truncal muscles was also noted. Superficial reflexes were absent and all deep tendon reflexes were exaggerated with bilateral ankle clonus and positive Babinski sign. Neither of the parents had similar skin lesions.
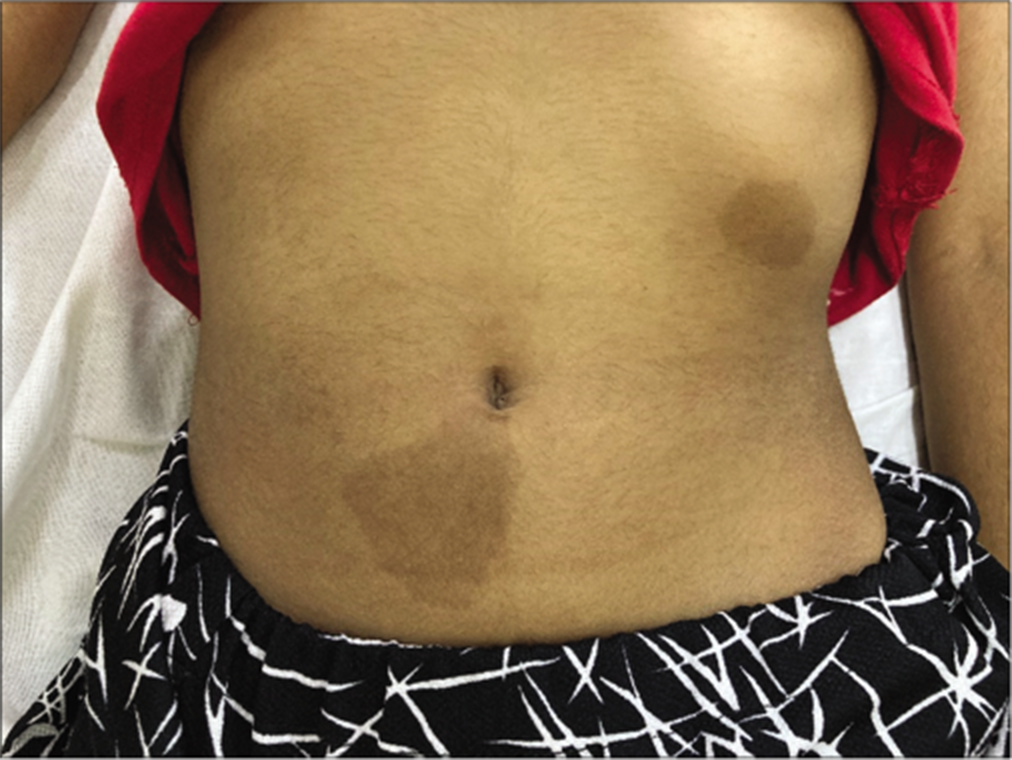
- Café au lait spots.
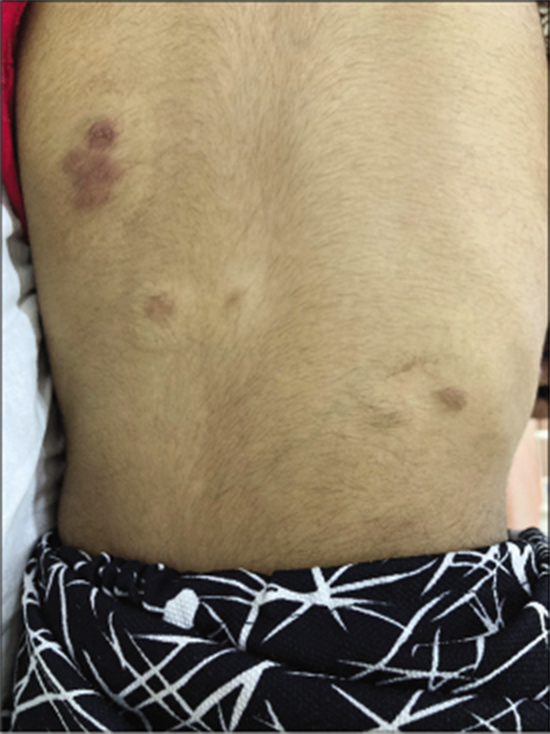
- Plexiform neurofibroma.
Based on the history and examination, a compressive myelopathy was suspected most likely to be a space occupying lesion of extramedullary origin. Due to the presence of neurocutaneous markers, neurofibromatosis was suspected. Magnetic resonance imaging (MRI) brain and spine was done which showed bilateral vestibular schwannoma with multiple neurofibromas along nerve roots in cervical and thoracolumbar areas located in the intradural extramedullary space [Figure 3]. There was a large neurofibroma at the cervicomedullary junction causing compression of the cervical cord. The tumor at the base of the skull was surgically excised and sent for histopathologic examination which revealed two distinct tumor morphologies, predominantly schwannoma with meningioma. Thus, a diagnosis of NF2 was made. The patient had poor prognosis due to presence of multiple neurofibromas along the entire length of the spinal cord.
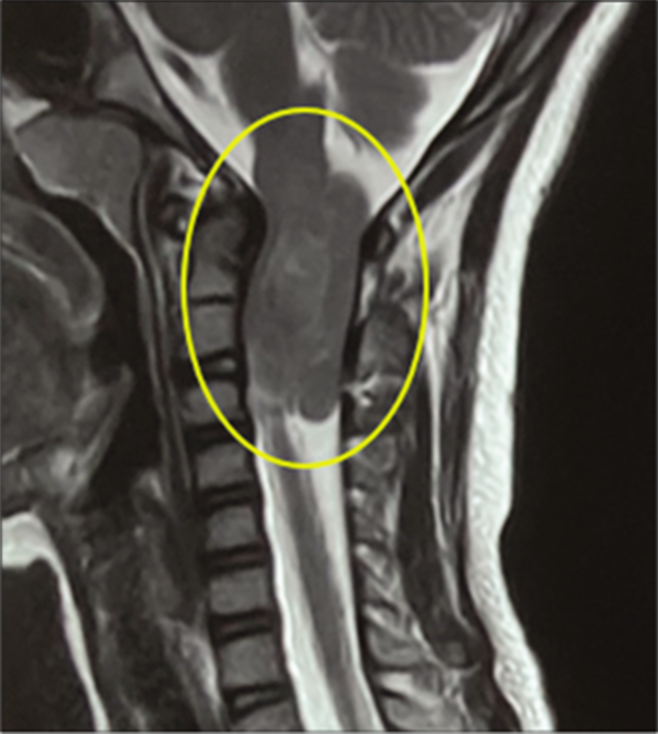
- Neurofibroma at cervicomedullary junction.
DISCUSSION
Compressive Myelopathies are classified as extradural and intradural pathologies which could be congenital (cysts), infective (abscesses, granulomas) and tumoral (benign/5 malignant). Extradural tumors may be benign like synovial cysts, osteomas, hemangiomas, etc. or malignant like bone metastasis, multiple myeloma, lymphoma. Intradural tumors can be further classified as extramedullary like neurofibromas, meningiomas, lipomas, schwannomas and arachnoid cysts and intramedullary which include astrocytoma, ependymoma, hemangioblastoma and metastasis.[3]
Extramedullary tumors usually present with UMN type of paralysis, have asymmetrical weakness which progresses in an inverted U-shaped pattern and late bowel bladder involvement. Intramedullary tumors present with LMN type pf paralysis, with symmetrical descending paralysis with bowel bladder involvement at presentation. Thus, our patient had features consistent with extramedullary tumor.
The Manchester (NIH) criteria (1992) is found to be highly sensitive for identifying cases of NF2 and is therefore used for the diagnosis.[4] It is as follows:
Bilateral vestibular schwannomas, or
-
Family history of NF2 plus
Unilateral vestibular schwannoma
Any 2 of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities.
Additional criteria
Unilateral vestibular schwannoma plus any two of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
Multiple meningioma (two or more) plus unilateral vestibular schwannoma or any two of: glioma, neurofibroma, schwannoma and cataract
The MRI of the above patient showed bilateral vestibular schwannoma and histopathology of the excised spinal lesions showed schwannoma and meningioma. Thus, the patient was found to satisfy the diagnostic criteria for NF2. This is one of the youngest reported cases of NF2.
There are no FDA approved drugs for treatment of NF Type 2 and surgical resection of tumors remains the mainstay of treatment. However, some new drugs are under evaluation for their role in shrinking tumor size. These include Bevacizumab (anti VEGF monocloncal antibody), Axitinib (VEGFR 1/2/3 inhibitor), Endostatin (VEGF expression inhibitor), Aspirin (Cox 2 inhibitor), Everolimus (inhibitor of mTORC1), Crizotinib (FAK1 inhibitor), Selumetinib (MEK1/2 inhibitor) and Lapatinib (EGFR/ ErbB2 inhibitor).[5]
The prognosis is guarded and depends upon the age at detection, the number and location of tumours. With newer therapies, survival rates are improving but overall prognosis continues to be poor with high rates of early mortality.
CONCLUSION
This case is rare because of the early age of presentation and absence of family history of similar lesions and cutaneous markers. It is important to follow a structured clinical approach in the localization of spinal space occupying lesions. Detailed clinical history, family history and examination are important to pick up soft markers of various diseases especially neuro cutaneous syndromes.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- WHO Classification of Tumors of the Central Nervous System Lyon: IARC/World Health Organization; 2016. p. :294-303.
- [Google Scholar]
- An update on the CNS manifestations of neurofibromatosis Type 2. Acta Neuropathol. 2020;139:643-65.
- [CrossRef] [Google Scholar]
- Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59:1759-65.
- [CrossRef] [PubMed] [Google Scholar]
- New developments in neurofibromatosis Type 2 and vestibular schwannoma. Neurooncol Adv. 2020;3:vdaa153.
- [CrossRef] [PubMed] [Google Scholar]




