
Translate this page into:
Inflammatory myofibroblastic tumor of intestine presenting as intussusception
*Corresponding author: Arka Banerjee, Department of Pediatric Surgery, Bai Jerbai Wadia Hospital for Children, Mumbai, Maharashtra, India. arkabanerjee6989@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Reddy KA, Banerjee A, Munghate G, Bendre PS. Inflammatory myofibroblastic tumor of intestine presenting as intussusception. Wadia J Women Child Health 2022;1(1):21-3.
Abstract
Inflammatory myofibroblastic tumor (IMFT) is a rare mesenchymal tumor predominantly seen in the lungs. Intestinal IMFT is extremely rare and may cause intussusception. Surgery is the mainstay of treatment as IMFTs are resistant to conventional chemoradiation. Long term follow up is required as there is high recurrence risk. About 50% of IMFTs have translocations involving the anaplastic lymphoma kinase (ALK) gene which has been therapeutically targeted using Crizotinib. We report a 1-year-old boy with ileo-ileal intussusception who was operated after a trial of conservative management. Laparotomy revealed an ileal mass that was resected and histopathology confirmed IMFT. Recovery was uneventful.
Keywords
Intestinal inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor
Intussusception

INTRODUCTION
Inflammatory myofibroblastic tumor (IMFT) is a rare mesenchymal tumor that usually presents in children and young adults.[1] Also known as inflammatory pseudotumor, plasma cell granuloma, inflammatory myofibrohistiocytic tumor and inflammatory fibrosarcoma, it is most commonly seen in the lungs. Rarer locations include small and large bowel mesentery, omentum, diaphragm, small and large bowel, appendix and abdominal wall. Of all the extrapulmonary sites, mesentery and omentum are the most common.[2]
We report a 1-year-old boy with IMFT of intestine presenting as intussusception.
CASE REPORT
A 1-year-old boy presented with a history of abdominal pain, non-bilious vomiting and passage of red currant jelly stools for 3 days. Clinically, there was visible intestinal peristalsis in the upper abdominal quadrants and a lump was palpable in the right iliac fossa. Ultrasonography revealed an ileo-ileal intussusception. Hence, he was initially treated conservatively but his symptoms didn’t subside. A computed tomography (CT) scan of the abdomen was done after 72 h of conservative management when the symptoms (and the lump) didn’t abate. It showed persistence of the ileo-ileal intussusception (in the proximal ileal loops, for a length of 1.4 cm) but did not highlight any lead points [Figure 1].
![CT scan (axial and sagittal images) of the abdomen showing ileo-ileal intussusception (bowel within bowel appearance) [arrow]](/content/147/2022/1/1/img/WJWCH-1-021-g001.png)
- CT scan (axial and sagittal images) of the abdomen showing ileo-ileal intussusception (bowel within bowel appearance) [arrow]
The child was taken up for an exploratory laparotomy and we found a hard stricturous mass in the ileum, approximately 50 cm proximal to the ileocecal junction [Figure 2]. The entire mesentery had enlarged lymph nodes. The strictured ileal segment was resected and sent for histopathological examination. Intestinal continuity was restored with an endto-end ileo-ileal anastomosis. Post-operative recovery was uneventful and the child was discharged 7 days after the surgery. The child is currently doing well 6 weeks from his surgery and does not have any complications.
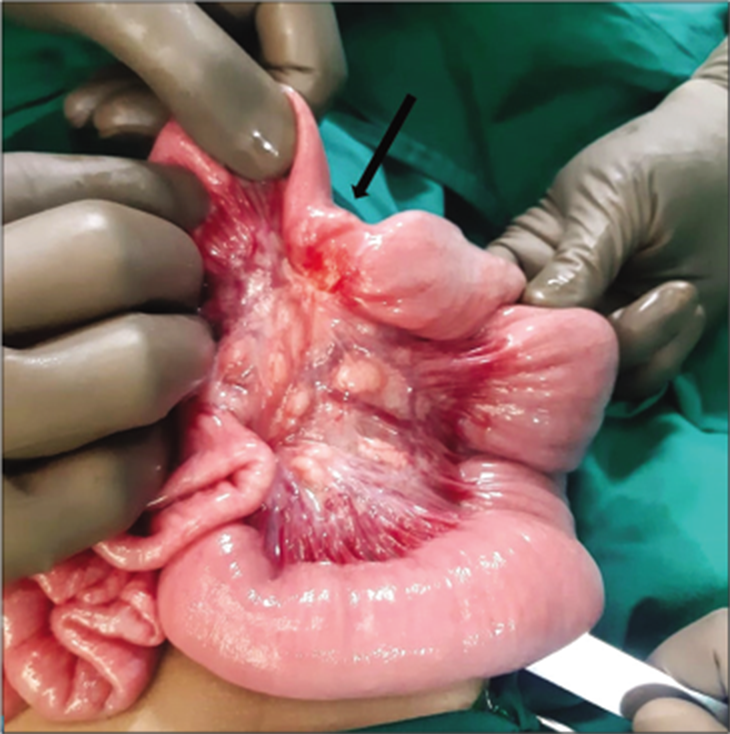
- Intra-operative picture showing a small fibrotic mass (arrow) causing ileal stricture.
Histopathology showed a spindle cell neoplasm involving the full thickness of the bowel wall with the resection margins free of the tumor. The tumour cells expressed SMA (Smooth Muscle Actin) and ROS-1 and were immunonegative for ALK-1 (Anaplastic Lymphoma Kinase – 1), CD34, S-100 protein and desmin on the basis of which a diagnosis of IMFT was made.
DISCUSSION
Predominantly seen in the lungs and airway and rarely in the intestine, IMFT is an intermediate soft tissue tumor that is composed of myofibroblast-differentiated spindle cells accompanied by numerous inflammatory cells, plasma cells and/ or lymphocytes. The exact etiology is unknown. Some believe it to be the result of an immunologic response to an infectious agent while others characterize it as a true neoplasm.[3]
Signs and symptoms depend upon the site of tumor. Abdominal mass, abdominal pain and sometimes intestinal obstruction are the common presentations of intra-abdominal tumors. IMFTs have a variable appearance on CT scan. They have well defined margins and may contain calcified and fatty compounds. As they are resistant to conventional chemotherapy and radiation, surgical excision is the mainstay of treatment.[4]
The tumor carries a risk of local recurrence after initial surgery (18–44%)[3] but has a very low risk of distant metastasis.[2] Abdominal and pelvic IMFTs have a recurrence rate of approximately 85%.[2]
The main genetic lesions underlying IMFTs are fusions involving the anaplastic lymphoma kinase (ALK) gene[5] and about half of the patients with IMFTs have translocations involving ALK.[6] The presence of specific genetic alterations in IMFT provides a strong rationale to therapeutically target ALK in this disease and Crizotinib, a small tyrosine kinase inhibitor targeting ALK, MET, ROS proto-oncogene 1 receptor tyrosine kinase (ROS1) and RON (Recepteur d’Origine Nantais), has been used to that effect with good results.[7] Corticosteroids, NSAIDs and radiotherapy have shown good results in ALK negative IMFT where surgery is not feasible.[3]
CONCLUSION
Intra-abdominal IMFT is a rare entity and can act as the lead point of intussusception in small children. Surgical excision is the mainstay of treatment and should be undertaken promptly in case of failure of conservative management, especially in atypical age group for developing intussusception. Histopathological analysis clinches the diagnosis. Long term follow up is required as there is a risk of recurrence.
Acknowledgment
We would like to thank Dr Foram Gala and Dr Hirva Manek from the Department of Radiodiagnosis, B J Wadia Hospital for Children for arriving at an accurate radiological diagnosis prompting us to explore the patient at the earliest.
We also extend our heartfelt gratitude to Dr Namrata and the entire pathology department at Raheja Hospital, Mumbai for their assistance with immunohistochemistry in this case.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Recurrent inflammatory pseudotumors in children. J Pediatr Surg. 2003;38:1491-5.
- [CrossRef] [Google Scholar]
- Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509-20.
- [CrossRef] [PubMed] [Google Scholar]
- Jejunal inflammatory myofibroblastic tumor: A rare entity. Int Surg J. 2017;4:2095-7.
- [CrossRef] [Google Scholar]
- Inflammatory myofibroblastic tumor of the small intestine. J Am Coll Surg. 2002;194:502-6.
- [CrossRef] [Google Scholar]
- Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A children's oncology group study. J Clin Oncol. 2017;35:3215.
- [CrossRef] [PubMed] [Google Scholar]
- An unusual case of systemic inflammatory myofibroblastic tumor with successful treatment with ALK-inhibitor. Case Rep Pathol. 2014;2014:470340.
- [CrossRef] [PubMed] [Google Scholar]
- Crizotinib: A comprehensive review. South Asian J Cancer. 2013;2:91.
- [CrossRef] [PubMed] [Google Scholar]




