
Translate this page into:
Clinical and radiological profile of a neonate with craniofacial microsomia – A case snippet
*Corresponding author: Sruthi Nair, Department of Neonatology, Seth GSMC and KEM Hospital, Mumbai, Maharashtra, India. sruthis.doc@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Prashanth RR, Nair S, Haribalakrishna A, Tekwani R, Clinical and radiological profile of a neonate with craniofacial microsomia – A case snippet. Wadia J Women Child Health. 2024;3(1):40-2. doi: 10.25259/WJWCH_50_2023
Abstract
Craniofacial microsomia (CFM) refers to a wide variety of phenotypic presentations resulting from underdevelopment of the mandible, maxilla, ear, orbit, facial soft tissue, and/or facial nerve. We report a case of CFM presenting with microtia, hemifacial microsomia, and limbal dermoid. In this case report, we describe the clinical course, investigations, and initial management of a neonate with CFM.
Keywords
Craniofacial microsomia
Limbal dermoid
Ear anomalies
Neonate
Microtia

INTRODUCTION
Craniofacial malformation results from underdevelopment of the mandible, maxillae, ear and soft tissue of the face. Previously referred to as Goldenhar syndrome these infants, in addition to abnormal development of facial features, also have additional features of vertebral and ophthalmic abnormalities. Early recognition and continued multidisciplinary care go a long way in the management of these infants.
CASE REPORT
A 37-week-old male neonate weighing 2620 grams was born, a non-consanguineous couple with two previous children, through lower segment cesarean section. Antenatal sonography including anomaly scan were reported normal. Baby cried immediately after birth and APGAR score was 7 at 1 and 5 min of birth. On examination, neonate was noted to have a distinct facial profile. There was hemifacial microsomia on the right side and deviation of the angle of the mouth to the left side with the normal right nasolabial fold suggestive of congenital absence of right angular oris. He was also noted to have a long philtrum, high arch palate, and arachnodactyly. Detailed examination of the ear showed microtia on the right side with the absence of the right tympanic membrane and the left ear had abnormal pinna. Eye examination revealed temporal limbal dermoid in the right eye with iris heterochromia, and rest of the right and left eye examination was normal [Figure 1]. The head circumference was 35 cm and length was 49 cm. Both were within normal limits. Systemic examination was normal. There was no family history of facial abnormality or maternal teratogenic drug intake.
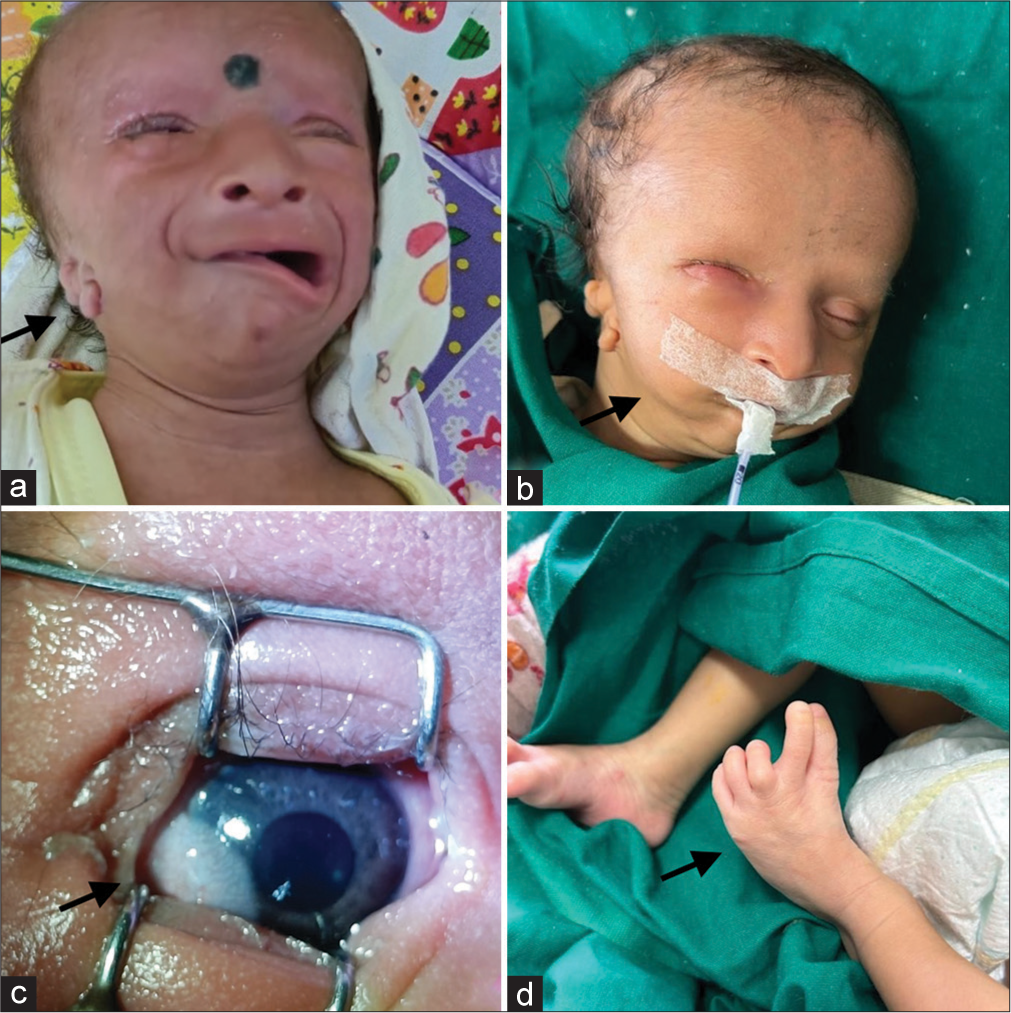
- Clinical features (a) hemifacial microsomia with right ear microtia (black arrow), (b) right sided mandibular hypoplasia (black arrow) with absence of right depressor angular oris, (c) limbal dermoid in right eye (black arrow) (d) arachnodactyly of left foot (black arrow).
The baby was further evaluated by a multidisciplinary team of a plastic surgeon, occupational therapist, lactation counselor, clinical psychologist, and social worker, and a thorough assessment of clinical dysmorphism by a geneticist showed no other abnormalities. Karyotyping was 46XY. There was no evidence of vertebral anomalies on X-ray. Thyroid function test was normal. Computed tomography of the bitemporal lobe revealed the absence of the right external auditory canal (EAC) and posterior semicircular canal. Dysplasia of malleus and incus were noted along with non-visualization of the long process of the incus. Cochlea showed two and a half turns. The internal auditory canal and bony facial canal were normal, with an intact temporomandibular joint. Similar findings were noted on the left side, with the exception of bony atresia of only the medial half of EAC and normal middle ear ossicles [Figure 2]. Ultrasound of the abdomen and echocardiography did not show any abnormality.
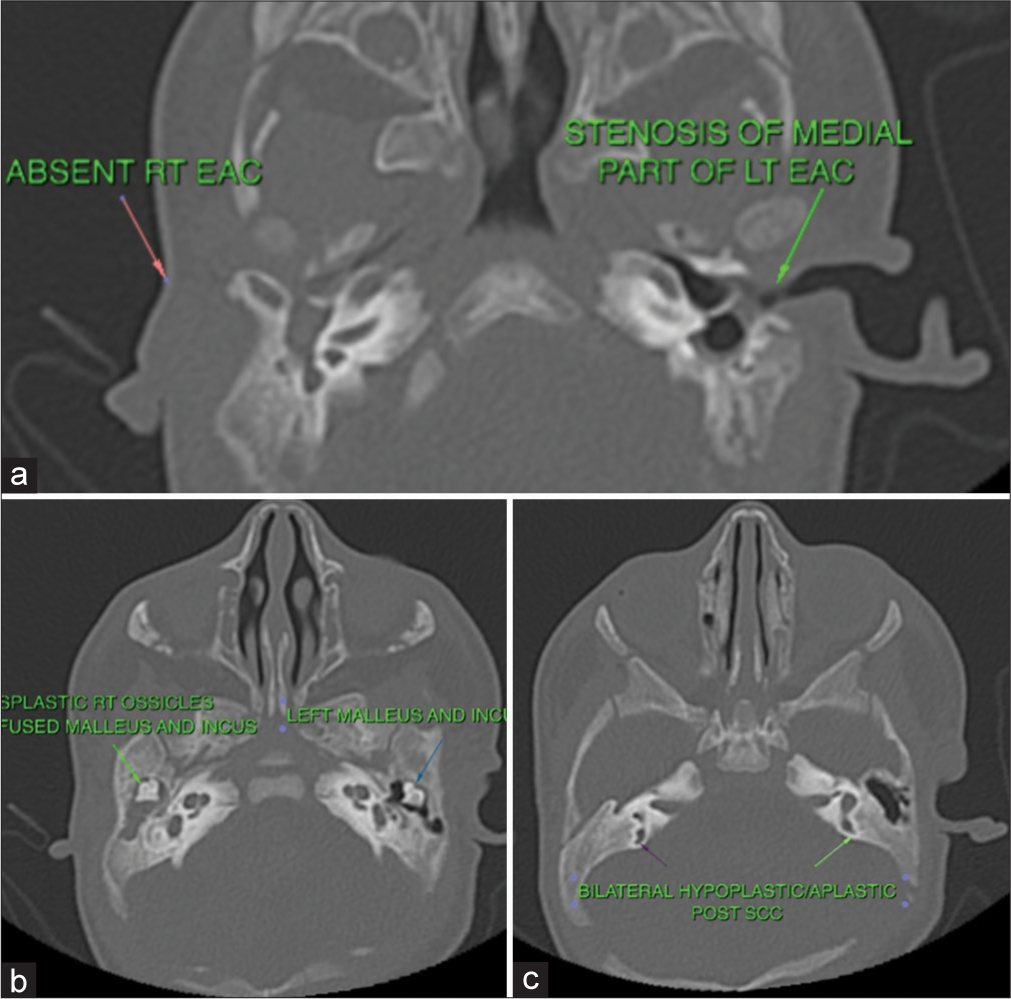
- Radiological features (a) bilateral hypoplastic external auditory canal (EAC), (b) dysplasia of middle ear bones, and (c) bilateral hypoplastic semi-circular canals (SCC).
Based on clinical and radiological findings, a diagnosis of craniofacial microsomia (CFM) was made and the baby was started on early intervention including visual and auditory stimulation. During the neonatal intensive care stay, he was given oral feeds through cup and spoon and was initiated on breastfeeding on 3rd day of life. Oromotor function was not impaired, and sucking and breastfeeding abilities were not disturbed. Oromotor exercises aided in easy transition to breastfeeds. There was no drooling of saliva or milk noted during feeds. Unsightly appearance, however, continued to be a concern for the mother.
Neurological assessment at the time of discharge was normal. A multidisciplinary team counseled the parents about the need for continued oral assessment for drooling, speech therapy, and corrective cosmetic surgery. The child was discharged on day 5 of life and advised fortnightly follow-up. Staged cosmetic correction and auditory rehabilitation is planned at 3 months of age.
DISCUSSION
Otomandibular dysostosis, oculoauriculovertebral syndrome, first and second branchial arch syndromes, and hemifacial microsomia are the various terms used in the literature to describe the abnormalities arising from the underdevelopment of facial features that arise from embryonic first and second pharyngeal arches.[1]
CFM refers to a wide variety of phenotypic presentations resulting from underdevelopment of the mandible, maxilla, ear, orbit, facial soft tissue, and/or facial nerve[2] and occurs in as many as 1/3000–1/5600 live births.[3]
CFM occurs due to disruption of the cellular, tissue to tissue communication that is essential for normal development of neuromuscular and skeletal components of the first and second pharyngeal arches. This disruption could be secondary to a vascular insult, teratogen exposure, and genetic causes. Autosomal dominance with incomplete penetrance is well described in the literature.
Pruzansky classification system has described the underdevelopment mandible in three groups based on radiographic features.[4] Kaban and associates later modified this system to describe the position of the temporomandibular joint.[5] Our patient had a grade 1 hemifacial microsomia with a small but otherwise normal mandible.
Bilateral involvement as in this neonate increases the risk of extracranial involvement in up to 35% and puts the neonate at additional risk of airway and feeding abnormalities.[6] Previously, the extracranial findings such as vertebral abnormalities and epibulbar dermoids were considered to be part of the Goldenhar syndrome but many clinicians recommend discontinuing the use of this term and using the terminology of CFM instead.[7]
These infants require long-term follow-up and staged reconstruction of face and auditory rehabilitation including cochlear implantation, ear reconstruction or prosthesis, aural atresia repair, jaw surgery, and soft-tissue augmentation starting from 3 months of age until 13–16 years of age.[8]
These infants also need continued multidisciplinary care to address the various problems related to oral health, growth, nutrition, and psychosocial well-being. Treatment of underdeveloped mandibles is complex and includes lengthening with bone distraction devices. Reduced oral intake due to limited oral opening due to mandibular hypoplasia, dental caries, or gingivitis requires to be addressed during each follow-up.[9] These children must also be followed up for behavioral problems, lower social competency, and lesser peer acceptance and provided psychosocial counseling until adolescence.[10]
CONCLUSION
CFM is a rare disorder requiring early recognition and diagnosis. The problems related to feeding need to be anticipated in the neonatal period and early intervention, auditory rehabilitation and facial reconstruction need to planned for better quality of life.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- The aetiology and pathogenesis of craniofacial deformity. Development. 1988;103:207-12.
- [CrossRef] [PubMed] [Google Scholar]
- Clarifying the relationships among the different features of the OMENS+ classification in craniofacial microsomia. Plast Reconstr Surg. 2015;135:149e-56.
- [CrossRef] [PubMed] [Google Scholar]
- Surgical correction of hemifacial microsomia in the growing child. Plast Reconstr Surg. 1988;82:9-19.
- [CrossRef] [PubMed] [Google Scholar]
- Craniofacial and extracraniofacial anomalies in craniofacial microsomia: A multicenter study of 755 patients. J Craniomaxillofac Surg. 2017;45:1302-10.
- [CrossRef] [PubMed] [Google Scholar]
- Goldenhar syndrome-ophthalmologist's perspective. Rom J Ophthalmol. 2018;62:96-104.
- [CrossRef] [Google Scholar]
- Mandibular microsurgical reconstruction in patients with hemifacial microsomia. Plast Reconstr Surg. 2008;122:1839-4.
- [CrossRef] [PubMed] [Google Scholar]
- Dental management of a child with Goldenhar syndrome. Eur J Gen Dent. 2014;3:158-62.
- [CrossRef] [Google Scholar]
- Psychosocial outcomes in children with hemifacial microsomia. J Pediatr Psychol. 2011;36:794-805.
- [CrossRef] [PubMed] [Google Scholar]




