
Translate this page into:
Childhood cystic lung diseases – A pictorial review
*Corresponding author: Dr. Alpa Bharati, Department of Radiology, Bai Jerbai Wadia Hospital for Children, Mumbai, Maharashtra, India. alpabharati@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Bharati A, Gala F, Manek H, Chandane P, Andharia JP. Childhood cystic lung diseases – A pictorial review. Wadia J Women Child Health 2022;1(1):51-7.
Abstract
Cystic changes in the lungs in neonatal age group as well as children are commonly encountered in day-to-day practice of paediatric chest imaging. It is therefore important to know the patterns of cystic disease, many of which are classical with definitive treatment options, to enable appropriate clinical care. This article briefly reviews the common conditions presented in our patient population with cystic changes in the lungs. The lesions have be classified as Congenital or autoimmune and Acquired conditions. The acquired lesions are largely infective in nature and represent a wide variety of infectious conditions.
Keywords
Cystic lung diseases
Children
CT scan

CONGENITAL CYSTIC LUNG DISEASES
Congenital diaphragmatic hernia (CDH)
These occur due to non-fusion of the pleuroperitoneal canals resulting in defect in the diaphragm. There is resultant intra-thoracic herniation of the abdominal contents mainly stomach, liver, intestines or spleen. CHD are categorised into two types based on anatomic location. CDH are associated with various congenital malformations including cardiac anomalies, intestinal malrotation, aneuploidy, sequestration and lung hypoplasia as well as neural tube defects.
Bochdalek’s Hernia is usually left sided and posterior or postero-lateral in location. It is the more common hernia and often large with poorer outcomes [Figure 1a and b].

- (a) Bochdalek’s hernia – axial high-resolution computed tomography image shows heterogenous solid cystic structure in the left hemithorax, more posteriorly located (Black arrow). A tubular structure also seen within is the section of the upward curled nasogastric tube (arrowhead). The heterogenous appearance is caused by air and gastric contents. (b) Bochdalek’s hernia – coronal view of the thorax in soft tissue setting of same patient as Figure 1 shows herniation of stomach (White arrow) along with the upward curve of the nasogastric tube (arrowhead).
Morgagni’s Hernia is less common and anterior in location. It may only show herniation of fat or part of liver along the mediastinum and may be an incidental finding [Figure 2a and b].
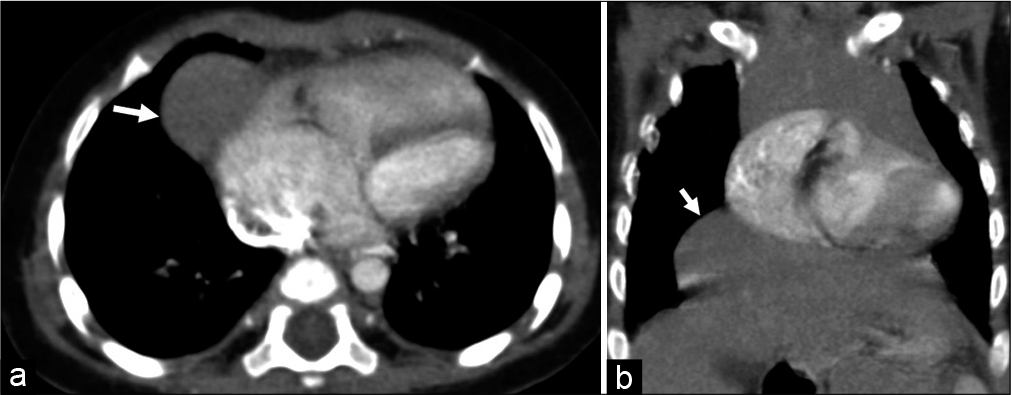
- (a) Morgagni’s hernia – a soft tissue lesion (arrow) is seen adjoining the right heart in a case of tetralogy of Fallot suggestive of herniation of liver parenchyma. (b) A well-defined soft tissue lesion in the right lower hemithorax, flush with the mediastinum is the superior herniation of the right lobe of liver (arrow) in the same case as in Figure 2b and represents a Morgagni’s hernia detected incidentally in a case of tetralogy of Fallot.
Antenatal ultrasound (US) and foetal magnetic resonance imaging (MRI) are useful tools for diagnosis of these conditions. Antenatal lung-head ratio calculated on ultrasonography or Fetal MRI can be used for prognostication.[1]
BRONCHOPULMONARY FOREGUT MALFORMATIONS
Congenital cystic adenomatoid malformation (CCAM)
It is also referred to as congenital pulmonary airway malformation and is a form of congenital bronchopulmonary foregut malformations. It occurs due to abnormal proliferation of the bronchoalveolar segments to form an adenomatous hamartoma having variably sized cystic areas. The lesions may also communicate with the bronchial tree. The lesions develop antenatally and can be detected on antenatal US and further evaluated on foetal MRI.
These lesions can become severely enlarged and compress the adjoining structures which may sometimes result in cardiac failure or maternal hydrops. Most however can be treated surgically in the neonatal period and are not life-threatening if appropriate timely care is provided [Figure 3a and b].
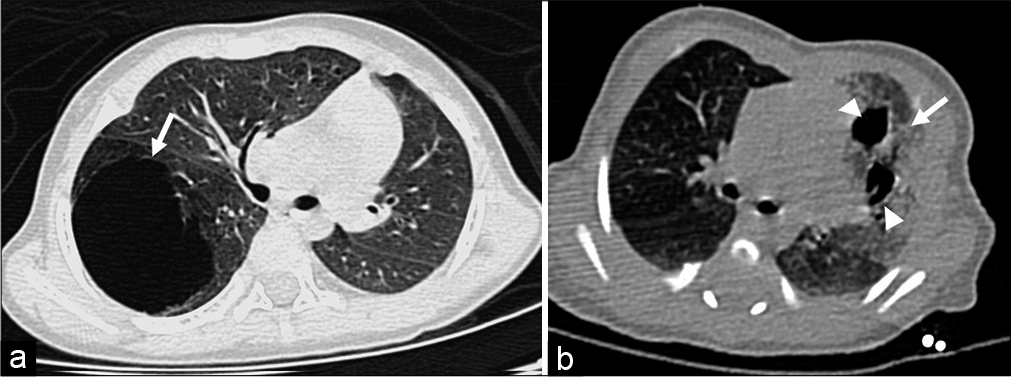
- (a) Congenital cystic adenomatoid malformation (CCAM) Type 1–high-resolution computed tomography (HRCT) axial image of a 1 month old male child shows a large air-filled cystic lesion in the right lower lobe. (b) CCAM Type 2. The HRCT image of neonate shows two well-defined somewhat irregular air-filled cyst (arrowheads) surrounded by hazy increased density lung parenchyma (arrow). The latter represents multiple tiny cysts coming together to give a hyperdense attenuation.
Pathologically CCAMs are classified into sub-types based on the size of the cysts.
Type I consists of large dominant macrocysts which may be surrounded by smaller microcysts. Type I CCAM is the commonest and represents almost 70% of the cases of CCAM[2]
Type II consists of cysts which are <2 cm and are often associated with other anomalies like renal agenesis, pulmonary sequestration or cardiac anomalies
Type III has sub-centimetre sized microcysts and often involves one entire lobe
Type IV often not distinguishable on computed tomography (CT) imaging from type I cyst. These pathologically have no cellular lining of the cyst[3]
Type 0 is lethal and represents global arrested lung development.[4]
Duplication cyst
These bronchopulmonary foregut malformations may be of bronchial, enteric or neurenteric subtypes.[5] The bronchial duplication cysts are lined by secretory epithelium and usually do not communicate with the bronchial tree. The cysts are therefore fluid filled and rarely show air within. The cystic fluid is often high in density and rarely may appear solid. The cyst contains varying amount of proteinaceous fluid, blood products and calcium oxalate[6] that can cause dependent areas of hyperdensity. The lesions are well defined, non-enhancing and densities ranging from fluid to soft tissue density depending on the composition of the fluid content. Almost 50% however will have fluid-density on CT[6] with attenuation values upto 20 Hounsfield Unit (HU). They are located close to the bronchial tree. Most of these are located in the hilar, subcarinal or right paratracheal regions. Less commonly the lesion may be intra-parenchymal. The latter are often in the peri-hilar region and in the lower lobe. If the lesions are large they can cause mass effect on the adjoining bronchus leading to obstructive emphysema. The lesions are seen as well-defined oval or rounded fluid density structures and show no enhancement on post-contrast imaging. The cysts may get complicated due to haemorrhage or secondary infection or fistula formation with the adjoining bronchus [Figure 4a-d]
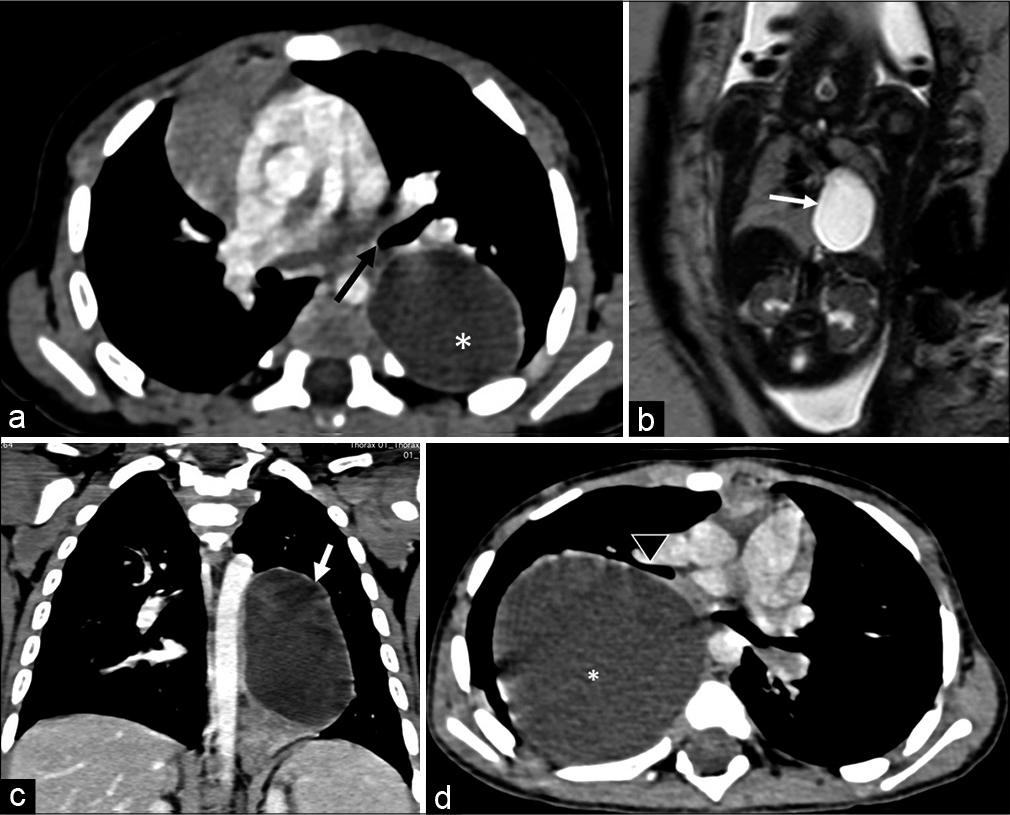
- (a) Bronchopulmonary foregut duplication cyst. it is seen in this post-contrast axial computed tomography (CT) scan of the chest as a hypodense and non-enhancing lesion (*). It is also seen close to the left lower lobe bronchus (orange arrow). (b) Foetal magnetic resonance imaging coronal plane: it shows a well-defined cystic intra-thoracic lesion (arrow) in the left hemithorax suspected of foregut duplication cyst. (c) Coronal CT image of the same patient as 4b shows the bronchopulmonary foregut duplication cyst in the left hemithorax (arrow). The cyst is single well defined and non-enhancing. It was also closely associated with the adjoining bronchus. (d) Large bronchogenic cyst (*) in the right hemithorax, closely related to the right middle lobe bronchus anteriorly (arrowhead). The cyst is fluid-density, well-defined and non-enhancing.
Sequestration
Pulmonary sequestration is also a bronchopulmonary foregut malformation that results in accessory lung tissue with no communication to the bronchial tree or pulmonary arterial system. It derives its blood supply from a systemic artery and drains into the pulmonary or systemic/azygous veins. This can cause a left–right shunt or high output cardiac failure. It has been classified as intralobar, presenting later in life with recurrent infections and extralobar presenting in newborns and infants with respiratory distress or infections.
The intra-lobar sequestered lesion does not have a separate pleura while the extralobar lesions are encased in their own pleura. Both these are usually seen in the lower lobes, more on the left side. Some extralobar lesions can be sub-diaphragmatic in location. On CT imaging, a systemic arterial supply to a soft tissue lesion can be demonstrated as well as its venous drainage. The lesions are well-defined, solid soft tissue density and have no air within unless complicated by infections. Lung sequestration is often associated with CCAM, CDH, congenital heart disease and scimitar syndrome[7] [Figure 5a-c].
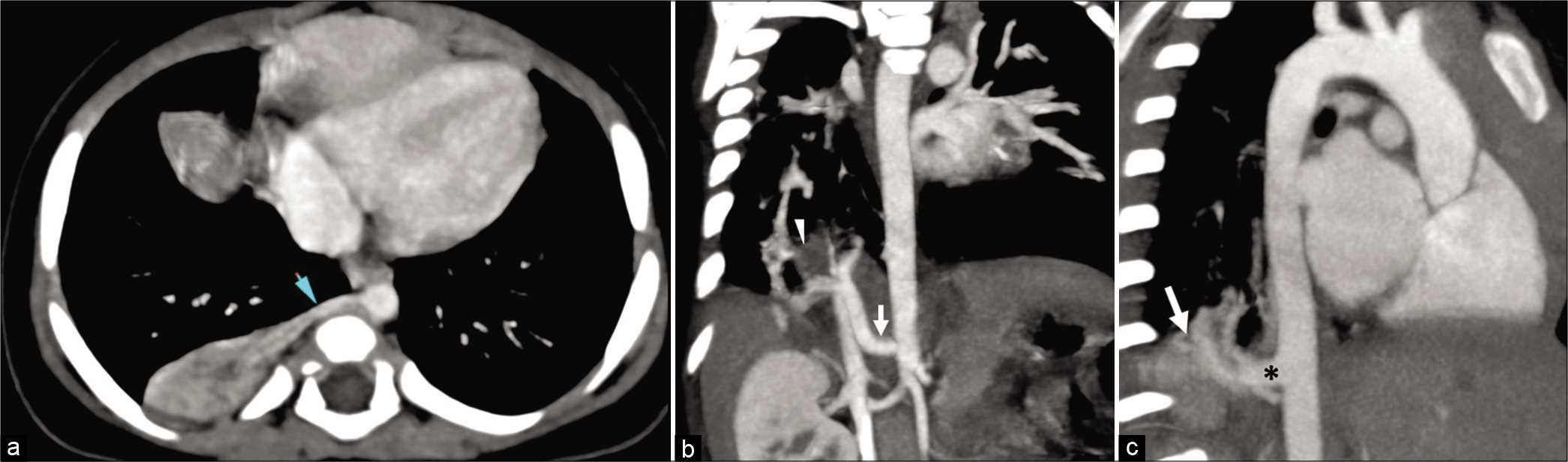
- (a) Extra-lobar sequestration. Axial post-contrast computed tomography (CT) image shows a well-defined solid lesion posterior to the right lower lobe arterial supply from the aorta (blue arrow). The lesion is well demarcated from the adjoining lung parenchyma. (b) Intralobar sequestration in a case with partial anomalous pulmonary venous return. Oblique coronal CT angiography image shows ill-defined hypodense area (arrowhead) in the right lower lobe being supplied by a large arterial branch from the abdominal aorta (arrow). The lesion shows irregular margins and not distinct from the adjoining ling parenchyma. (c) Another case of intra-lobar pulmonary sequestration shows irregular isodense lesion along the right lower lobe with prominent arterial supply from the aorta.
CHILDHOOD INTERSTITIAL LUNG DISEASE
Langerhans cell histiocytosis (LCH)
It is a multisystem disease of variable presentation and prognosis. It may be disseminated with multisystem affection (Letterer -Siwe disease), multiple lesions but single system affection (Hand – Scheuller-Chrisitian disease) or a solitary lesion in one organ (eosinophilic granuloma). Bones are important sites of manifestation of all three above manifestation of the disease.
Lungs are often involved in multisystem disease. It typically involves the upper and middle lobes. The earliest manifestations are centrilobular nodules of about 1cm or less in size. The nodules undergo cavitation forming thick-walled cysts and later evolve to thin walled cysts. Cysts are late manifestation of the disease. Often the adjoining cysts merge to form large cysts of bizarre shapes. The intervening lung parenchyma is normal in early stage of the disease but later shows ground glass opacities, mosaic attenuation and septal thickening. Later in the disease fibrosis sets in with larger cysts and honeycombing.
Complications of the disease include pneumothorax, pneumomediastinum, pulmonary hypertension and cor pulmonale and respiratory failure [Figure 6a-c].
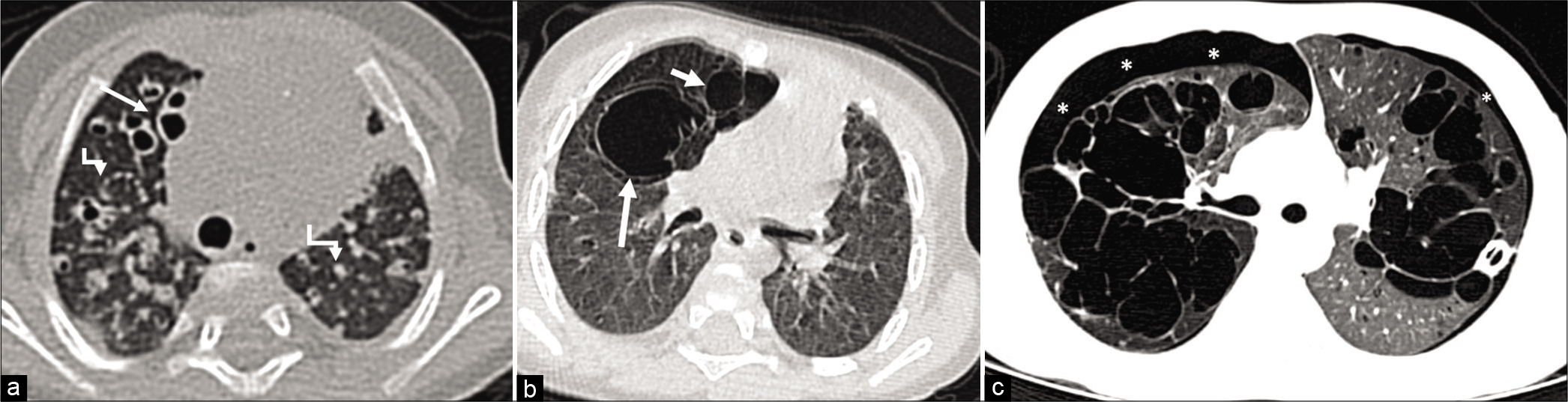
- (a) Langherhans Cell Histiocytosis-high-resolution computed tomography axial image of a 2 month old child shows multiple solid nodules (bent arrows) in the bilateral lung parenchyma with many show cystic changes. Some of the cysts in the right middle lobe are large in size (arrow). (b) Same patients in 6a post-chemotherapy at age 9 months shows larger bizarre shaped cysts in the right middle lobe (arrows) and resolution of the previous nodular lesions. (c) Another case of LCH. Both lungs show variably sized and bizarre shaped cysts in the lung parenchyma. Also seen in bilateral pneumothorax (Asterix), a known common complication of LCH. The left pneumothorax is slight seen as a fine around the left lung.
Cystic bronchiectasis
Many conditions both genetic and acquired may be associated with cystic bronchiectasis. Cystic Fibrosis and ciliary motility disorders are well-known genetic conditions associated with cystic bronchiectasis. Besides these aspiration pneumonias, allergic bronchopulmonary aspergillosis and immunodeficiency with recurrent pneumonias can also present on imaging with cystic bronchiectatic changes. Bronchiectasis is associated with inflammation of the bronchial wall followed by bronchial dilatation and distortion. The high-resolution CT findings include bronchial wall thickening and dilatation of the bronchi which may be seen extending to the peripheral one third of the lung parenchyma. The bronchial dilatation causes a ‘signet-ring’ appearance where the bronchial lumen is seen like a ring with a normal sized accompanying bronchiole as a signet over it. The dilated bronchi may be occluded by thick mucus plugs and seen as linear branching hyperdense areas. Surrounding nodular infiltrates are typically seen in presence of on-going infection. The disease can be localised in acquired cases and diffuse in genetic conditions. There is however a lower lobe predilection in all cases [Figure 7a-c].
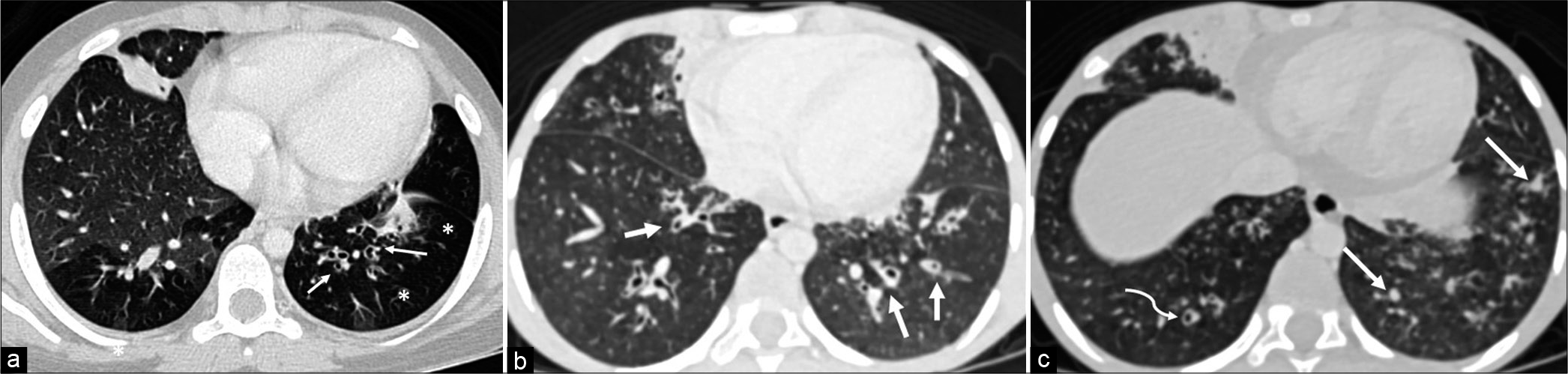
- (a) Cystic Bronchiectasis. The high-resolution computed tomography (HRCT) axial image shows early bronchiectasis changes in the left lower lobe. There is signet-ring sign with the bronchial size being larger than the accompanying arteriole (arrows). Bronchiectasis is often associated with mucoid plugging of the bronchioles and thereby cause obstructive emphysema of the distal lung (asterisk) is hypodense and shows reduced vascularity compared to the normal lung. (b) Cystic Bronchiectasis. The HRCT axial image shows bronchiectatic changes in the both lings. Concentric bronchial wall thickening and bronchial dilatation is seen (arrows). Also noted are centrilobular nodular infiltrates suggesting infection. (c) The HRCT axial image of the same patient in 7b shows mucous plugging of the terminal bronchioles (arrow) as hyperdense areas. Curved arrow points to the classic signet-ring appearance of bronchiectasis.
Other childhood interstitial lung disease are also associated with cystic lesions including in the neonatal period. Most of these show associated ground glass opacities and septal thickening with variable distribution patterns. Of note is granulomatosis with polyangiitis (Wegener’s Granulomatosis) which is often seen with variably sized nodules having central cavitation. The cavities have thick irregular walls. Bronchopulmonary dysplasia is another condition which may show cystic appearance due to air- trapping along with septal and bronchial wall thickening and mosaic lung attenuation.
INFECTIOUS DISEASES
Pulmonary tuberculosis
Cystic changes in pulmonary tuberculosis are well known and are almost always seen in post primary tuberculosis. Almost 20–45% of post-primary pulmonary tuberculous disease show cavitation, usually in the posterior segment of the upper lobes[8,9] but can be extensive and patchy. These are usually a result of cavitation of the caseating granulomas, lung parenchymal destruction and bronchiectatic changes. The cystic areas are irregular air-filled and of variable size. The lesions are often found in later stage of the disease with fibrotic changes. Often the surrounding lung shows centrilobular nodules forming a tree-in-bud pattern due to endobronchial spread of disease.[8,10] The cavities are usually thick-walled and may show air-fluid levels within. Other accompanying findings include enlarged necrotic mediastinal and hilar nodes, pleural effusions or empyema and lung consolidations [Figures 8a and b].
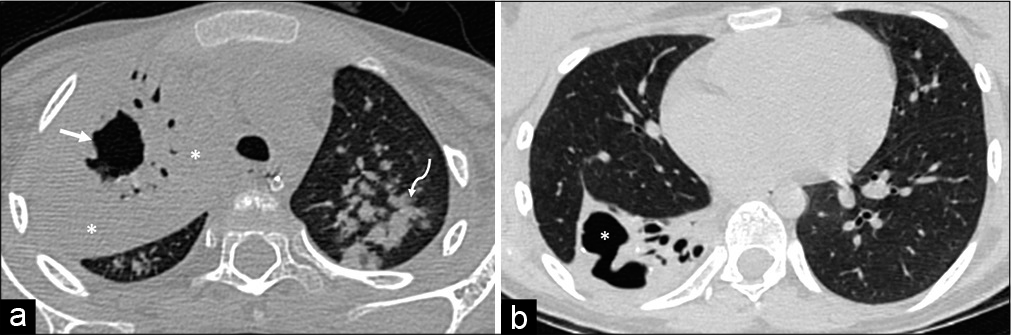
- (a) Pulmonary Tuberculosis-An irregular cystic lesion is seen in the right upper lobe (arrow) with surrounding dense lung consolidation (*). There are smaller cystic areas seen around it are sections dilated bronchi. Also noted are confluent nodular infiltrates in the left lung (curved arrow). (b) Superior segment of right upper lobe is collapsed with areas of bronchiectasis and large irregular shaped air-filled cystic lesion (*). There is volume loss and parenchymal destruction of the superior segment of right upper lobe.
CYSTIC LESIONS ASSOCIATED WITH NONMYCOBACTERIAL PNEUMONIAS
Lung necrosis pneumatoceles and abscess
Non-mycobacterial pneumonias can cause pneumatoceles or can result in necrosis of the consolidated lung. The former are commonly seen in staphylococcal pneumonia and the latter is often seen in streptococcal pneumonia usually in combination of either Haemophilus influenzae or Pseudomonas aeruginosa.[11]
Lung necrosis
Necrotising pneumonia is a serious complication of community acquired pneumonia. It results in pulmonary inflammation, consolidation followed by necrosis and formation of small cavities.[12] The necrotic areas are seen as hypodense non-enhancing regions within a consolidated lung segment. These are often seen communicating with the adjoining pleural space resulting in empyema. Necrotic areas are filled with fluid-density material and may contain air specks or air-fluid level suggestive of bronchial communication [Figure 9a and b].
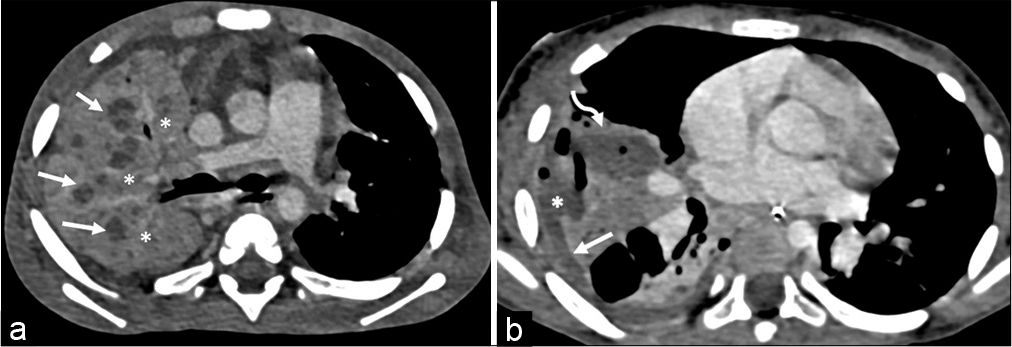
- (a) Necrotising pneumonia. There is extensive dense consolidation of the right lung parenchyma with multiple hypodense non-enhancing areas within representing areas of lung necrosis (arrows). Consolidated lung is diffusely hyperdense (*). (b) Necrotising pneumonia with empyema. There hypodense non-enhancing (curved arrow) within the consolidated right lung appearing hypodense. These are showing communication (*) with adjoining pleural space with resultants small empyema (arrow). Few discrete air-pockets are also seen within the necrotic foci and pleural space.
Pneumatoceles
Are usually post-infectious sequalae though in neonates they can occur secondary to ventilator induced lung injury. The commonly involved bacterial agents include Staphylococcus aureus, Streptococcus pneumoniae, H. influenzae, Escherichia coli and P. aeruginosa pneumoniae. Pneumatoceles are air-filled well defined lesions usually asymptomatic. These can get secondary infection and result in clinical symptoms. Sometimes the pneumatoceles can become large and compress the adjoining lung parenchyma. These can rupture and cause spontaneous pneumothorax [Figures 10a and b].
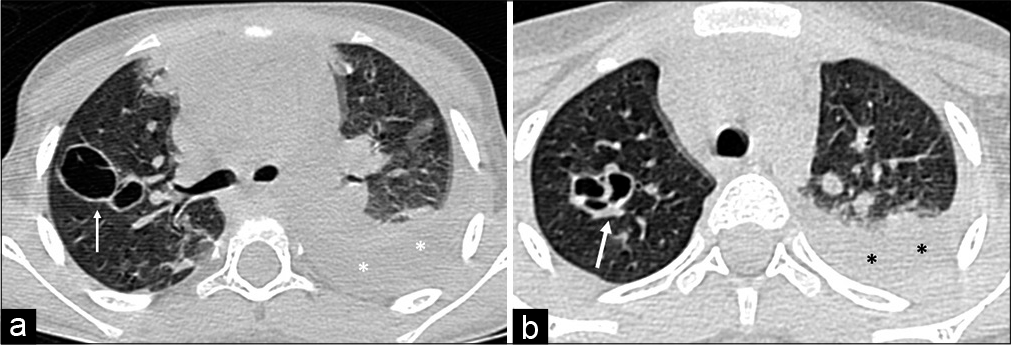
- (a and b) Pneumatoceles in a case of drug resistant staphylococcal pneumonia. The pneumatoceles (arrow) are thin walled variably sized air-filled lesions. Also present is left sided effusion with underlying collapse consolidation (*).
Abscess
Bacterial and mycobacterial infections can result in lung abscess. The abscess can be primary from primary lung infection as in cases of aspiration or necrotising pneumonia or tuberculosis.
Secondary abscess may develop in an existing lung condition such as in foreign body aspiration or bacterial endocarditis. Abscess is seen on CT imaging as a variably sized round or oval areas of lung necrosis and liquefaction appearing fluid in density. The central fluid-density is non-enhancing and is surrounded by an thick enhancing wall. Adjoining lung is usually consolidated. The consolidation may resolve with treatment much earlier than the abscess.[13] Often bronchi and pulmonary vessels may be seen reaching to the abscess wall.[13] Air-fluid level and internal septations may be seen within the abscess. Most common organisms implicated in abscess include S. aureus, Klebsiella pneumoniae and Pseudomonas[13] [Figures 11a and b].
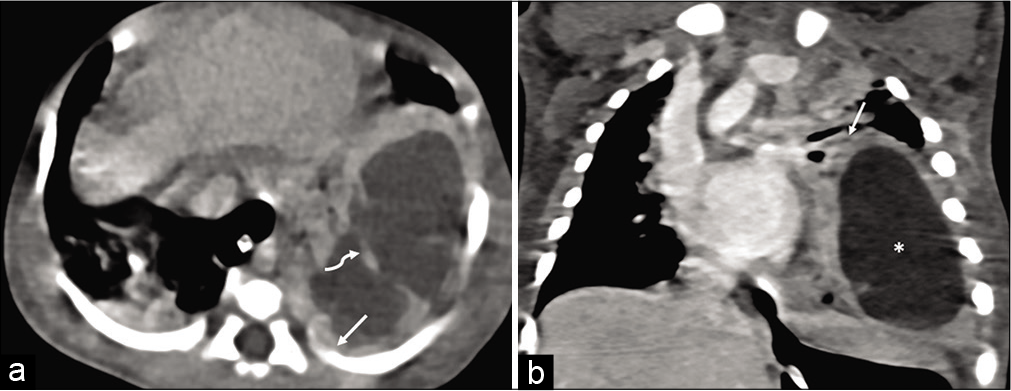
- (a) Lung abscess-axial computed tomography scan shows a large necrotic lesion with thick irregular walls (arrow) and internal septation (curved arrow) in the left hemithorax. There is rim of consolidated lung around the lesion (arrow in 10b). (b) Lung abscess. Coronal computed tomography image shows the abscess (*) and surrounding lung consolidation (arrow).
Septic emboli
These result from infectious thrombotic material which reaching the lung through the pulmonary arteries. It is often seen in patients with infective endocarditis (right sided)[14] or those with non-cardiac focus of infection and having intracardiac septal defects. Other sources include spread from infected indwelling venous lines and periodontal disease.[15,16] Almost always, the disease affects bilateral lung parenchyma. The septic infarcts are typically seen as multiple peripheral sub-pleural nodules with or without cavitation or necrosis. There may be associated surrounding lung consolidation. A feeding vessel sign has been described where a peripheral nodule is seen with a traceable adjoining feeding vessel[17] [Figures 12a and b].
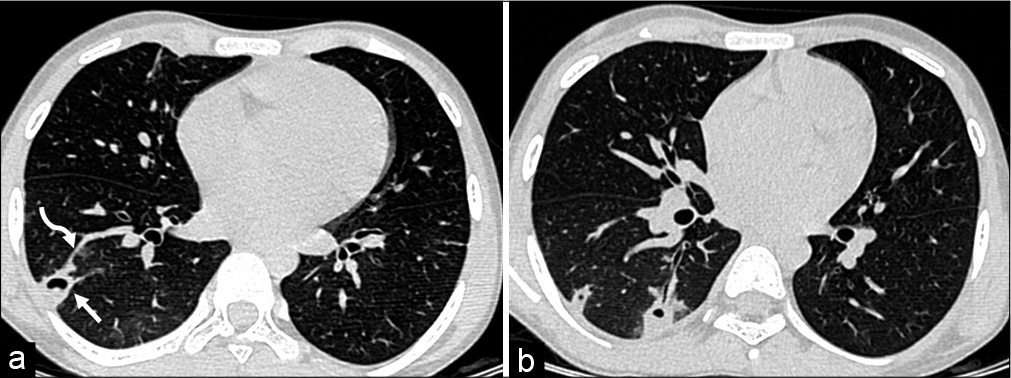
- (a) Septic emboli. A peripheral sub- pleural necrotic nodule (arrow) is seen with a vessel traceable to it (curved arrow). The patient is a case of VSD with septicemia. The lesion is typical of a septic embolus. (b) Septic emboli. Same patients as 11a. Few other similar lesions are seen in peripheral sub-pleural location.
Fungal pneumonia
Fungal infection can either cause cavitatory lesion or develop in a pre-existing cavitatory lesions such as in cavitatory tuberculosis. These are commonly seen in immunocompromised host including post-transplant patients.
Multiple nodular lesions with surrounding ground glass opacities are typical appearance of fungal lesions, commonly mucormycosis in post-transplant individuals. These nodules may show central areas of cavitation. Aspergillosis and mucormycosis both have a propensity for invading the pulmonary arteries causing infarction-haemorrhagecavitation. This cavitation with a rim of air results in the air-crescent sign and suggests onset of recovery.[18] The Monod sign on the other hand is typically seen in aspergillosis where it forms a fungal ball within an existing air-filled cavitatory lesion[19] [Figure 13a and b].
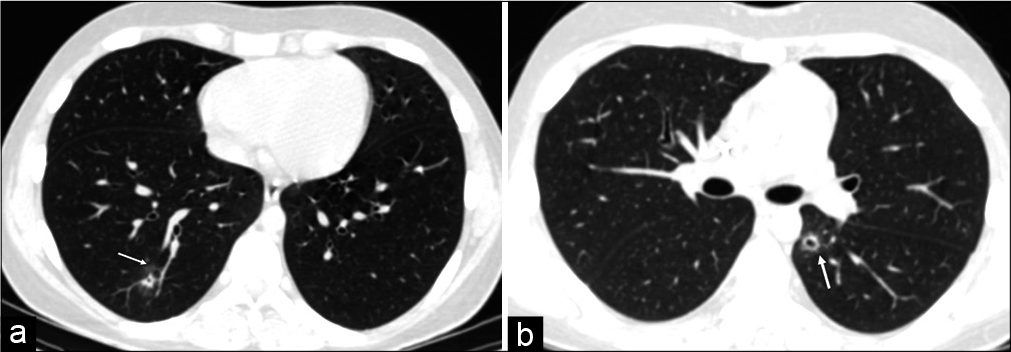
- (a) Fungal nodules with cavitation. High-resolution computed tomography (HRCT) axial image of a patient post bone marrow transplant shows a nodule in the posterior basal segment of right lower lobe (arrow) with small central cavitation seen as black spot. There is a faint of ground glass opacity (arrow) around the lesion typical of a fungal nodule. (b) Fungal nodule with cavitation. HRCT axial image of another post bone marrow transplant patient with known fungal infection, shows a cystic lesion (arrow) with a well-defined wall in the left lower lobe. It represents cavitation within the fungal nodule.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Congenital diaphragmatic hernia: Predictive value of MRI relative lung-to-head ratio compared with MRI fetal lung volume and sonographic lung-to-head ratio. Am J Roentgenol. 2009;192:153-8.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital lung abnormalities: Embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics. 2010;30:1721-38.
- [CrossRef] [PubMed] [Google Scholar]
- AIRP best cases in radiologic-pathologic correlation: Type 2 congenital cystic adenomatoid malformation (type 2 congenital pulmonary airway malformation) Radiographics. 2011;31:743-8.
- [CrossRef] [PubMed] [Google Scholar]
- Concurrent bronchopulmonary foregut malformations: A rare case of right-sided extralobar pulmonary sequestration and bronchogenic cyst. Egypt J Radiol Nucl Med. 2021;52:54.
- [CrossRef] [Google Scholar]
- Mediastinal bronchogenic cyst: Demonstration of a fluid-fluid level at MR imaging. Radiology. 1993;186:427-8.
- [CrossRef] [PubMed] [Google Scholar]
- Scimitar syndrome: A European congenital heart surgeons association (ECHSA) multicentric study. Circulation. 2010;122:1159-66.
- [CrossRef] [PubMed] [Google Scholar]
- Imaging of Pulmonary Infections Philadelphia, Pennsylvania: Lippincott Williams and Wilkins; 2007.
- [Google Scholar]
- Pulmonary tuberculosis: The essentials. Radiology. 1999;210:307-22.
- [CrossRef] [PubMed] [Google Scholar]
- Computed Tomography and Magnetic Resonance of the Thorax. Respiratory Care. 2008;53:1234.
- [Google Scholar]
- Thin-section CT findings of patients with acute Streptococcus pneumoniae pneumonia with and without concurrent infection. Br J Radiol. 2012;85:e357-64.
- [CrossRef] [PubMed] [Google Scholar]
- Surgical management of lung gangrene. Can Respir J. 2000;7:401-4.
- [CrossRef] [PubMed] [Google Scholar]
- Large septic pulmonary embolus complicating Streptococcus mutans pulmonary valve endocarditis. J Radiol Case Rep. 2018;12:18-27.
- [CrossRef] [PubMed] [Google Scholar]
- Septic pulmonary emboli as a complication of peripheral venous cannula insertion. Drug Discov Ther. 2018;12:111-3.
- [CrossRef] [PubMed] [Google Scholar]
- Septic pulmonary emboli due to periodontal disease. Respir Med. 2006;100:1470-4.
- [CrossRef] [PubMed] [Google Scholar]
- High-resolution MDCT of pulmonary septic embolism: Evaluation of the feeding vessel sign. Am J Roentgenol. 2006;187:623-9.
- [CrossRef] [PubMed] [Google Scholar]
- Maging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams and Wilkins; 2007.
- [Google Scholar]
- Varied radiologic appearances of pulmonary aspergillosis. Radiographics. 1995;15:1273-84.
- [CrossRef] [PubMed] [Google Scholar]




