
A case of infantile hypophosphatasia: Phenotypic findings of a compound heterozygous inheritance with a novel mutation
*Corresponding author: Anuradha V. Khadilkar, Department of Pediatrics and Growth and Pediatric Endocrinology, Hirabai Cowasji Jehangir Medical Research Institute, Jehangir Hospital, Pune, Maharashtra, India. anuradhavkhadilkar@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Dange NS, Mondkar SA, Khadilkar V, Khadilkar AV. A case of infantile hypophosphatasia: Phenotypic findings of a compound heterozygous inheritance with a novel mutation. Wadia J Women Child Health 2023;2(1):26-9.
Abstract
We report the case of a 9-month infant who presented with failure to thrive and bony deformities, mimicking a ricket-like picture. Biochemical parameters showed hypercalcemia, low serum alkaline phosphatase, normal serum phosphorus, magnesium, and parathormone levels. Ultrasound revealed nephrocalcinosis. Clinical exome sequencing revealed infantile hypophophatasia with two compound heterozygous mutations in exon 6 (mutation being novel) and 12. To the best of our knowledge, compound heterozygous mutations in exon 9 and exon 12 have presented with pyridoxine responsive seizure (PRS) along with hypophosphatasia (HPP). This is the third case of infantile HPP reported from India, but with a novel mutation who had not yet manifested with PRS.
Keywords
Infantile hypophosphatasia
Alkaline phosphatase liver gene
Tissue non-specific alkaline phosphatase
Metaphyseal mineralization defects

INTRODUCTION
Hypophosphatasia (HPP) is a rare disorder of bone metabolism occurring due to a loss of function mutation in the gene coding for tissue non-specific alkaline phosphatase (TNSALP) enzyme, inherited in either an autosomal dominant or recessive manner. It has a remarkably wide spectrum of severity ranging from in utero or neonatal death to isolated dental manifestations in adults. The perinatal and infantile forms are the most severe and carry a poor prognosis.[1]
HPP is characterized by defective mineralization of skeletal and dental structures. Clinical features, radiological findings, and/or perturbed biomarker levels (especially alkaline phosphatase [ALP]) should raise suspicion and point toward HPP as one of the differential diagnoses. Most commonly, these children present with skeletal deformities and diagnosis may be confused with rickets due to limited awareness about the disorder. Misdiagnosis and treatment with Vitamin D and/or calcium supplements only worsens the disorder. We report a rare case of infantile hypophophatasia presenting with failure to thrive and bony deformities, with compound heterozygous mutations, one of the mutations being novel, not yet reported in the literature.
CASE DETAILS
A 9-month-old boy, an only child, born to nonconsanguineous parents was referred to our tertiary level Pediatric Endocrinology center for failure to thrive and global developmental delay. He was born vaginally at term, with a birth weight of 3.2 kg and had an uneventful postnatal period. His mother first noticed his failure to gain weight at 5 months of age when he weighed 3.5 kg. Simultaneously, she also noticed global developmental delay (not achieved head holding, could only make cooing sounds) and feeding difficulties and found the infant to be floppy. He had no history of repeated vomiting, chronic diarrhea, polyuria, or seizures. He had a history of one admission at 7 months of age for a lower respiratory tract infection.
On examination, he was alert, but had failed to thrive (weight for height Z-score = −2.2), with a weight of 3.8 kg (−6.3 Z-score), length of 55 cm (−7.2 Z-score) and head circumference of 44 cm (−0.5 Z-score). He was dolichocephalic, with large open fontanelles and prominent eyes. He had signs of florid rickets, hypotonia and a bell-shaped chest, normal genitals, and no organomegaly [Figure 1]. In view of differential diagnoses of metabolic bone disease, inborn errors of metabolism and renal disorder, blood, and radiological investigations were performed which revealed that serum sodium, potassium, urea, creatinine, and venous blood gas analysis were within the reference range [Table 1]. The metabolic bone profile revealed hypercalcemia (ionic and total), serum phosphorous and magnesium concentrations were within the reference range, parathormone concentrations were suppressed and the urinary calcium: creatinine ratio was significantly elevated [illustrated in Table 1]. Extremely low serum ALP concentrations with severely under-mineralized bones (assessed radiologically) led to a suspicion of HPP. Ultrasound of the kidneys revealed medullary nephrocalcinosis. The child was prescribed a tablet of hydrochlorthiazide (3 mg/ kg/day in two divided doses), advised calcium restricted feeds, and samples were sent for clinical exome sequencing analysis. Genetic testing revealed compound heterozygous mutations in exon 12 (c.1403C>T [p.Ala468Val]) and exon 6 (c.493A>G [p.Thr165Ala]) of the ALP Liver gene (ALPL) confirming the diagnosis of “infantile HPP” [Figure 2]. He is slated for a follow-up after 3 months.
| Lab parameter | Value | Reference range |
|---|---|---|
| Haemoglobin | 8.8 g/dL | 11.1–14.1 g/dL |
| White blood cells count | 8550/cmm | 6000–16000/cmm |
| Platelet count | 350×103/cmm | 150–450×103/cmm |
| Creatinine | 0.3 mg/dL | 0.1–0.8 mg/dL |
| Calcium | 11.5 mg/dL | 9.0–11.0 mg/dL |
| Phosphorus | 5.7 mg/dL | 4.5–6.7 mg/dL |
| Alkaline phosphatase | 16.0 U/L | 150.0–350.0 U/L |
| Parathyroid hormone | 3.9 pg/mL | 13.6–85.5 pg/mL |
| Ionic calcium | 1.40 mmoL/L | 1.15–1.29 mmoL/L |
| Sodium | 136.0 mmoL/L | 138.0–145.0 mmoL/L |
| Potassium | 4.2 mmoL/L | 3.4–4.7 mmoL/L |
| Magnesium | 2.1 mg/dL | 1.7–2.3 mg/dL |
| Urine calcium/creatinine ratio | 1200.6 | 12–244 mg/g creatinine |
| Venous blood pH | 7.350 | 7.38–7.44 |
| Bicarbonate | 21.6 mmoL/L | 20.0–24.0 mmoL/L |

- A 9-month-old infant with dolichocephaly, bell shaped chest, Harrison’s sulcus, and failure to thrive diagnosed with infantile hypophosphatasia.
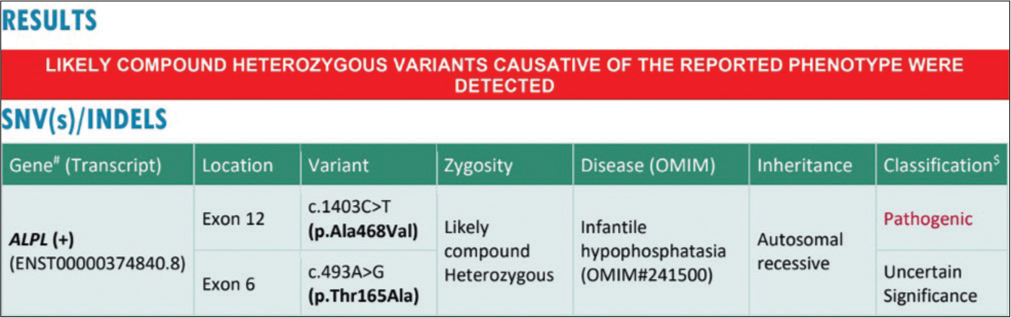
- Clinical exome sequence report.
DISCUSSION
A 9-month-old infant presenting with failure to thrive and a ricket-like picture had predominantly metaphyseal under-mineralization on radiographs along with biochemical parameters pointing toward HPP which was then seconded by clinical exome sequencing. HPP is a rare condition caused by mutations of the ALPL gene coding for TNSALP). Mutations in the ALPL gene are majorly found in bones, developing teeth and also in kidneys, lungs, and liver.[2] Autosomal recessive inheritance pattern with biallelic loss of function of ALPL (homozygotes or compound heterozygotes) is associated with severe phenotypes diagnosed in utero, whereas the autosomal dominant forms with heterozygous mutations are associated with milder phenotypes. HPP is classified based on age at symptom onset and severity, as a lethal perinatal, benign perinatal, infantile, juvenile, adult, and odonto form. The severity of HPP symptoms usually correlate s with residual ALP activity.[1] Infantile forms are considered less severe than perinatal forms and present before 6 months of age. Initial clinical presentation may be in the form of skeletal deformities with pseudorachitic metaphyseal broadening, curved limbs, and progressive sternal hollowing. Radiologically, generalized, and irregularly distributed hypomineralization of bone particularly in metaphyseal areas is seen. Such infants may also have feeding difficulties, axial, and peripheral hypotonia, failure to thrive or repeated rhinobronchitis.[3] Nearly half of the patients with infantile HPP develop early decidual tooth loss, recurrent respiratory tract infections, craniosynostosis, hypercalcemia, and/or nephrocalcinosis. Rarely, pyridoxine responsive seizures (PRS) are seen in patients diagnosed with infantile HPP because of accumulation of pyridoxal phosphate which is not dephosphorylated into pyridoxine and cannot cross the bloodbrain barrier. Vitamin B6-dependent seizures and respiratory failure are the leading causes of mortality in infantile HPP.[4]
Our patient presented with failure to thrive, developmental delay and a history of respiratory tract infection requiring hospitalization. Although clinically he had rachitic signs with metaphyseal bony changes, distinctive biochemical laboratory investigations raised suspicion of HPP highlighting the importance of investigations in severe rickets. Compound heterozygous mutations, that is, in exon 12 (c.1403C>T [p.Ala468Val]) and a novel mutation in exon 6 (c.493A>G [p.Thr165Ala]) of the ALPL gene were seen. This novel mutation in exon 6 is characterized by craniosynostosis, hypotonia, decreased TNSALP, infantile onset HPP, elevated urine and plasma inorganic pyrophosphate, elevated serum phosphate, failure to thrive, short stature, primary premature tooth loss, seizures, and developmental delay.[5] Both these variants have not been reported in the 1000 genomes and gnomAD.[6] Previously reported compound heterozygous mutations in exon 9 and exon 12 had presented as PRS along with HPP unlike that seen in our case with exon 6 mutation.[7,8] Although a literature search revealed many infantile HPP cases reported worldwide, only two cases have been reported from India based on clinical and biochemical suspicion.[9,10]
Cardinal laboratory parameters in infantile HPP are low ALP accompanied by hypercalcemia, hyperphosphatemia, and low parathyroid hormone concentrations. Hypercalciuria with or without elevated serum calcium concentrations result s in nephrocalcinosis.[2] Developmental delay in such patients is attributed to their skeletal defects and muscle weakness. Studies have demonstrated that the final height-adjusted mean/median bone mineral density (BMD) Z-scores in all groups of HPP patients are usually within the normal range. However, lower limb BMD may be affected as evidenced from lower-than-normal hip BMD Z-scores for age. Muscle weakness is also a known phenomenon as evidenced from weaker grip strength.[3]
Management of patients with HPP is largely symptomatic and multidisciplinary. Good ventilation strategies and respiratory physiotherapy are required in respiratory compromise. Nutritional and exogenous calcium/vitamin D sources are restricted to prevent hypercalcemia. Nasogastric nutrition or in severe cases, a gastrostomy may be considered in cases with feeding difficulties and failure to thrive. Bone deformities need meticulous follow-up and at times, orthopedic correction. Antiresorptive agents like bisphosphonates and denosumab and Vitamin D supplements should not be used to treat HPP. Enzyme replacement therapy has a promising effect on skeletal outcomes, pulmonary, and physical function in life-threatening HPP. Treatment with asfotase alfa has been shown to have an effective role with improved mineralization of skeleton, including the ribs, improved respiratory function, and improved survival with perinatal and infantile HPP.[11] Definitive treatment of infantile HPP is still difficult due to non-availability of asfotase alfa in India.
CONCLUSION
We found a novel mutation in our rare case of infantile hypophophatasia. In routine practice, consideration should be given to the rare differential diagnosis of HPP and radiographical and biochemical parameters may help in arriving at the diagnosis. This may avoid treating the misdiagnosed cases of HPP as rickets with Vitamin D supplements and worsening of the disorder.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Natural history of perinatal and infantile hypophosphatasia: A retrospective study. J Pediatr. 2019;209:116-24.e4.
- [CrossRef] [PubMed] [Google Scholar]
- Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C. p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007;40:1655-61.
- [CrossRef] [PubMed] [Google Scholar]
- McKusick's online mendelian inheritance in man (OMIM®) Nucleic Acids Res. 2009;37:D793-6.
- [CrossRef] [PubMed] [Google Scholar]
- Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patient with hypophosphatasia. Hum Mol Genet. 1994;3:1683-4.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile hypophosphatasia secondary to a novel compound heterozygous mutation presenting with pyridoxine-responsive seizures. JIMD Rep. 2013;11:17-24.
- [CrossRef] [PubMed] [Google Scholar]
- SUN-450 infantile hypophosphatasia-a novel mutation. Kidney Int Rep. 2020;5:S382.
- [CrossRef] [Google Scholar]
- Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016;101:334-42.
- [CrossRef] [PubMed] [Google Scholar]




