
Translate this page into:
Pyrexia of unknown origin with lymphadenopathy - An uncommon diagnosis
*Corresponding author: Sumitra Venkatesh Department of Pediatrics, Bai Jerbai Wadia Hospital for Children, Mumbai, Maharashtra, India. sumitravenkatesh99@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Meena S, Venkatesh S. Pyrexia of unknown origin with lymphadenopathy - An uncommon diagnosis. Wadia J Women Child Health. 2024;3(1):43-5. doi: 10.25259/WJWCH_22_2023
Abstract
Kikuchi disease or histiocytic necrotizing lymphadenitis is a benign and self-limited disorder characterized by tender regional cervical lymphadenopathy. We report a case of a 4-year 5-month old female presenting with fever and lymphadenopathy for 2 months. All infective and inflammatory cause of pyrexia of unknown origin was negative. Lymph node histopathology showed features of Kikuchi’s disease.
Keywords
Kikuchi necrotizing lymphadenitis
Systemic lupus erythematous
Karyorrhectic debris

INTRODUCTION
Kikuchi-Fujimoto disease (KFD) is a rare lymphohistiocytic disorder, first described in 1972 by Kikuchi and Fujimoto.[1,2] It is also known as Kikuchi disease or Kikuchi necrotizing lymphadenitis with predominant clinical features being fever and lymphadenopathy. KFD generally affects individuals between 20 and 35 years of age, with a male: female ratio of 1:2 and has a higher prevalence among Japanese and Asian populations. The exact cause of this disease is not clear. A viral or autoimmune etiology has been implicated. The role of Epstein– Barr virus as well as other viruses (HHV6, HHV8, Parvovirus B19) may also be considered in the pathogenesis of the disease. Some cases show histological findings analogous to that of systemic lupus erythematous (SLE). However, association of KFD with SLE remains unclear. It is characterized by tender cervical lymphadenopathy and flu-like symptoms. The constitutional symptoms such as prolonged fever and night sweats often lead to a mistaken clinical diagnosis of an infection or malignancy.
However, morphological aspects may mimic SLE, non-Hodgkin lymphoma, and reactive lymphadenopathy of other origins. The diagnosis of KFD is established by finding distinctive histological abnormalities in the lymph node.
CASE REPORT
A 4 year 5-month-old female, 1st by birth order, born of 3rd degree consanguineous union, presented with chief complaints of fever since 2 months. There was a history of two previous hospital admissions for similar complaints and on further enquiry there was a history of significant weight loss. There was no history of loss of appetite, joint pain or swelling, and bone pain/night pain or rash. There was no history of tuberculosis in the family or close contacts.
She had a normal antenatal and birth history with normal growth and development.
On examination, she had generalized lymphadenopathy-right cervical level II multiple lymph nodes, firm in consistency, left cervical level II lymph nodes 1.5 × 2 cm, mobile, and left cervical level III lymph nodes sub-centimetric; left axillary 1 × 0.5 cm and bilateral inguinal lymphadenopathy, sub-centimetric. Her height and weight were between −1 and −2 standard deviations scores (SDS). The systemic examination was unremarkable. Complete blood count revealed anemia with a normal leucocyte count. Inflammatory markers were significantly raised [Table 1]. There was no evidence suggestive of tuberculosis. Blood and urine cultures showed no growth. The peripheral smear showed microcytic and hypochromic anemia with neutrophil predominant white blood cells. The antinuclear antibody (ANA) test and ANA blot were negative. Two-dimensional-echocardiography evaluation was also normal.
| Parameters, unit | Observed values | Normal range |
|---|---|---|
| Hemoglobin, mg/dl | 7.9 | 11–14 |
| Total white blood cell count, c/mm3 | 7,500 | 5–15,000 |
| Platelet count, lakh/µl | 2.15 | 1.5–3.5 |
| C-reactive protein, mg/L | 48 | <5 |
| Erythrocyte sedimentation rate, mm/hr | 100 | 0–15 |
| Serum aspartate aminotransferase, U/L | 49 | <32 |
| Serum alanine aminotransferase, U/L | 42 | <33 |
| Lactate dehydrogenase, U/L | 494 | 120–300 |
| Serum uric acid, mg/dl | 1.9 | 2.4–5.7 |
| Complement component 3 (C3), mg/dl | 134 | 66–185 |
An ultrasonography of the neck showed bilateral cervical lymphadenopathy with necrotic nodes in the left cervical level IV with largest measuring 2.1 × 0.8 cm. The cervical lymph node was biopsied and sent for histopathology which revealed distorted architecture of lymph nodes composed of eosinophilic granular necrotic material and apoptotic bodies. There was expansion of small lymphocytes admixed with immunoblasts. No pathogen or tubercular granuloma was seen on histopathological examination. Overall features were suggestive of Kikuchi disease [Figures 1 and 2]. The child was given symptomatic treatment with antipyretics.
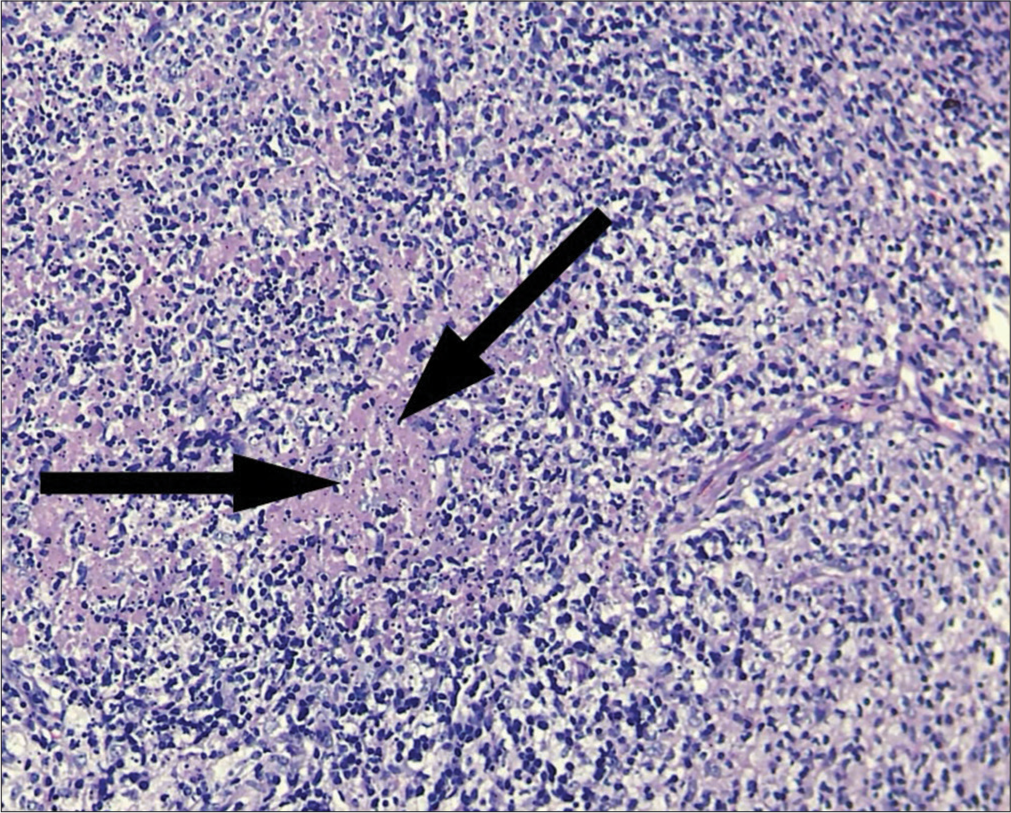
- Karyorrhectic debris (black arrows), histiocytes, fibrin deposits, and plasmacytoid monocytes.
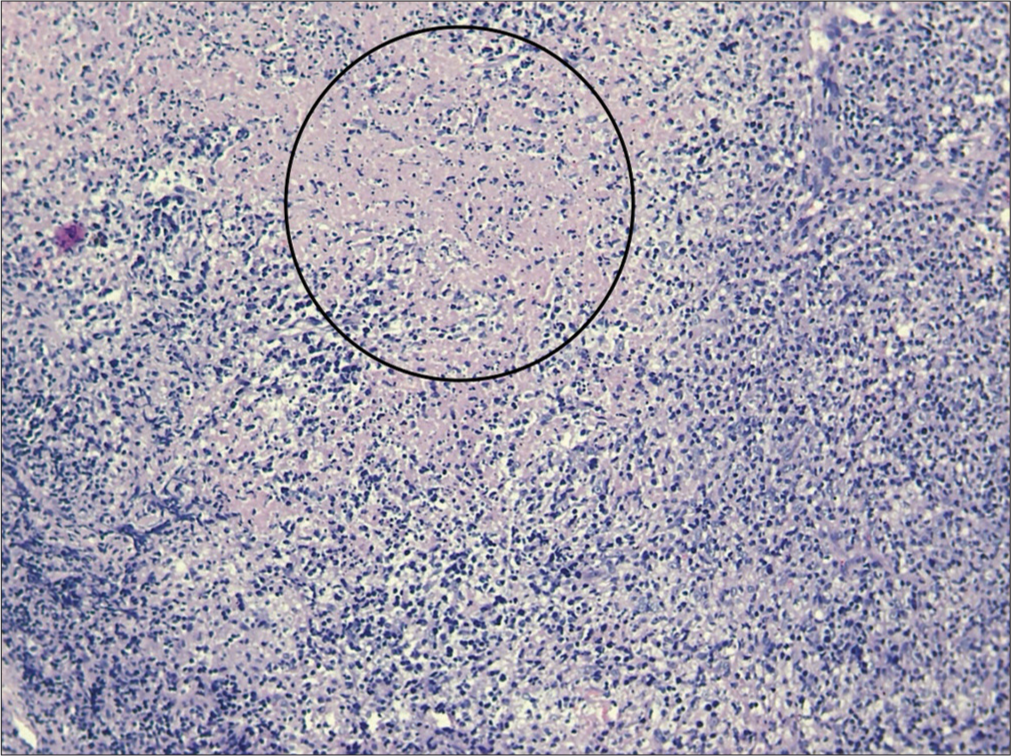
- Lymph node histopathology showing pale circumscribed foci of necrotic lesions (inside circle), giving starry sky appearance.
DISCUSSION
KFD is a rare, benign, self-limiting cause of fever, and cervical lymphadenopathy.[3] It has an acute or subacute onset evolving throughout 2–3 weeks. This disease typically involves cervical lymph nodes (mostly posterior cervical triangle). The size of lymph nodes is usually 1–4 cm; however, size as large as 8 cm have been reported. Other lymph nodes may also get involved. Symptoms may include associated fever, arthralgia, myalgia, sore throat, night sweats, weight loss, and hepatosplenomegaly. Involvement of extranodal sites is uncommon but skin, eyes, lung, pleura, joint, and bone marrow involvement have been reported. If there is extra nodal involvement, systemic symptoms are more likely to be noticed. However, in a country like India with a high prevalence of tuberculosis, the concomitant diagnosis of tuberculosis should be investigated. Other differential diagnosis include HIV disease, lymphoma, infectious mononucleosis, Kawasaki disease, toxoplasmosis, and cat scratch disease. Due to multisystem involvement and spontaneous resolution of lymphadenopathy, sarcoidosis can also be included in the differential diagnosis. No specific laboratory test is available for diagnosis.[4] A complete blood count is usually within normal range, though sometimes, neutropenia and atypical lymphocytosis may be seen.[5] Other laboratory abnormalities include elevated erythrocyte sedimentation rate, lactate dehydrogenase, and alanine aminotransferase. KFD is diagnosed by an excisional biopsy of the afflicted lymph nodes. The architecture of the involved lymph nodes is partially obscured by paracortical nodules of apoptotic necrosis that includes plenty of karyorrhectic debris and large numbers of histiocytes. The plasmacytoid monocytes tend to congregate particularly at the edges of necrotic foci. Numerous tiny lymphocytes and immunoblasts are intermingled together. Neutrophils and eosinophils are absent with scanty or absent plasma cells.
Immunohistochemistry reveals histiocyte associated antigens such as lysozyme, myeloperoxidase, and CD 68. In lymphocyte population, there is predominance of T-cells with very few B-cells and abundance of CD8 + T-cells over CD 4+T-cell. KFD is a self-limiting condition usually resolving within 1–4 months, with a recurrence rate of 3–4%.[6-8] Therapy is focused on symptomatic relief, including reduction of fever and lymph node tenderness with the use of analgesics and antipyretics. Corticosteroids are only used in the most severe or relapsing disease.
CONCLUSION
KFD is a rare benign cause of lymphadenopathy. In cases of lymphadenopathy with generalized symptoms of malaise, fever, and fatigue, it should always be taken into account as a differential diagnosis. Histopathology is crucial for a correct diagnosis because it can mimic tubercular lymphadenopathy or malignant lymphoma. Because patients may eventually develop lymphoproliferative malignancy or SLE, long-term follow-up is crucial.
Acknowledgements
The authors wish to acknowledge the Department of Histopathology, Bai Jerbai Wadia Hospital for Children.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Cervical subacute necrotizing lymphadenitis. New clinicopathologic entity. Naika. 1972;20:920-7.
- [Google Scholar]
- Kikuchi necrotising lymphadenitis in a child. Dubai Med J. 2020;3:126-38.
- [CrossRef] [Google Scholar]
- Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease. Cancer Control. 2014;21:313-21.
- [CrossRef] [PubMed] [Google Scholar]
- Kikuchi's disease (histiocytic necrotizing lymphadenitis): A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology, and DNA ploidy. Am J Surg Pathol. 1995;19:798-809.
- [CrossRef] [PubMed] [Google Scholar]
- Kikuchi-Fujimoto disease in the United States: Three case reports and review of the literature [Erratum appears in Mediterr J Hematol Infect Dis 2014;6:E2014023] Mediterr J Hematol Infect Dis. 2014;6:e2014001.
- [CrossRef] [PubMed] [Google Scholar]
- Enigmatic KikuchiFujimoto dis-ease: A comprehensive review. Am J Clin Pathol. 2004;122:141-52.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical presentations, laboratory results and outcomes of patients with Kikuchi's disease: Emphasis on the association between recurrent Kikuchi's disease and autoimmune diseases. J Microbiol Immunol Infect. 2010;43:366-71.
- [CrossRef] [PubMed] [Google Scholar]




